

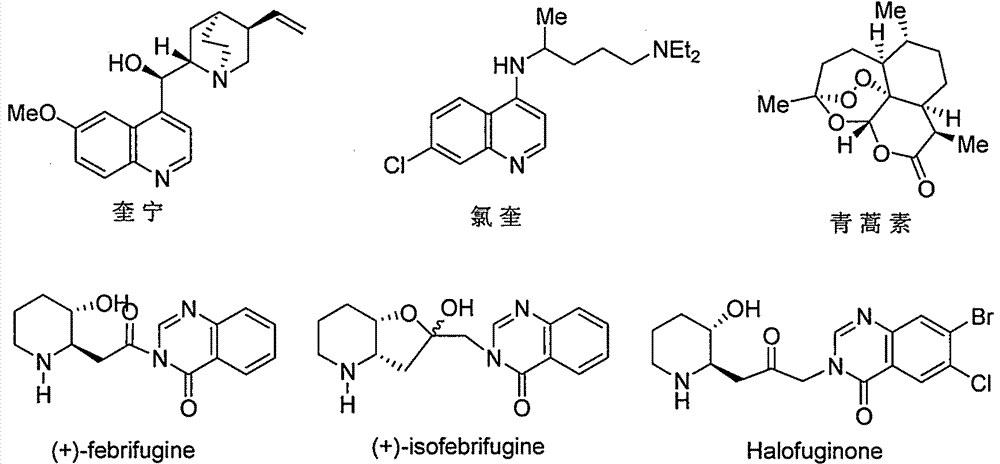
Preparation method of key chiral intermediate of febrifugine and halofuginone
A technology of chiral intermediates and hemosanone, which is applied in organic chemical methods, bulk chemical production, organic chemistry, etc., can solve the problems of cumbersome routes, expensive prices, and difficult operations, and achieve simple routes, simple operations, and reduced costs Effect
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

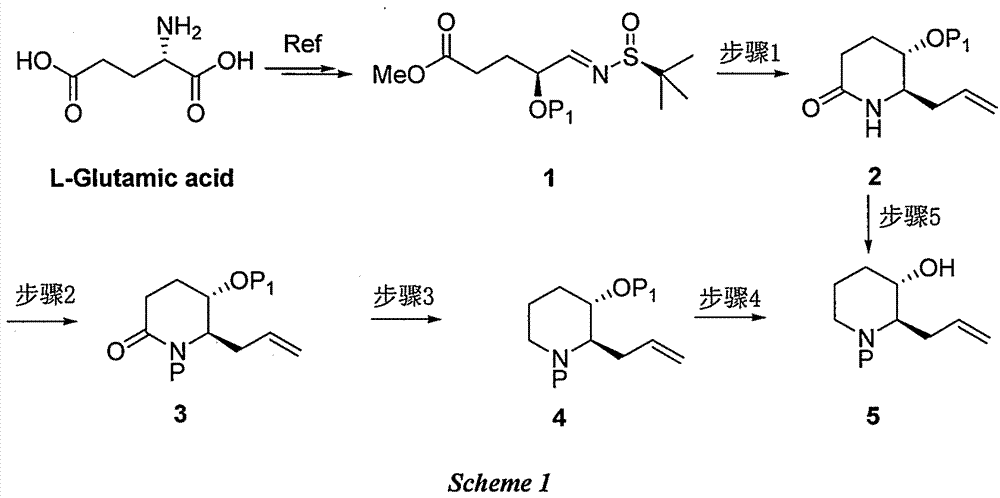
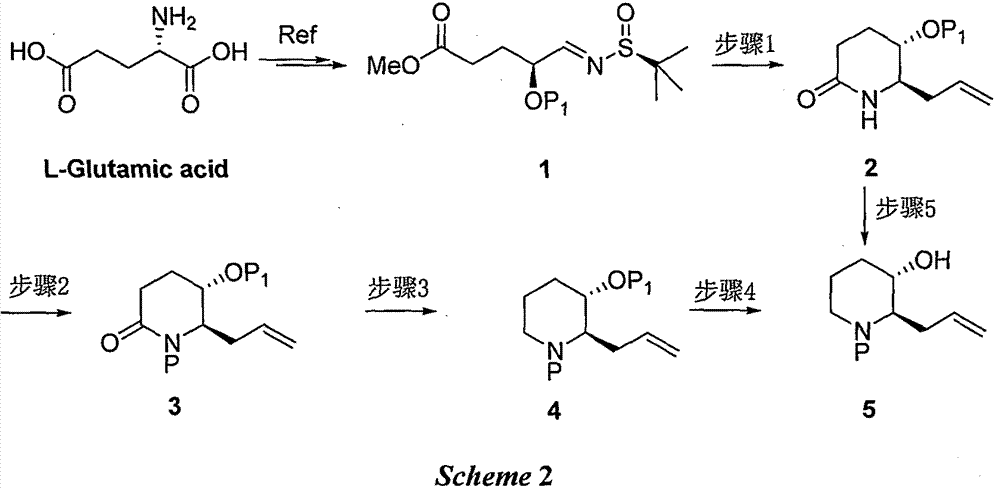
Examples
Embodiment 1
[0021] Step 1: Synthesis of (5S,6R)-6-allyl-5-(tert-butyldimethylsilyloxy)piperidin-2-one 2(P 1 =TBS)
[0022] Dissolve compound 1 (2.0g, 5.51mmol) in 50mL of water, add indium powder (1.27g, 11.02mmol), add allyl bromide (1.33g, 11.02mmol) dropwise after stirring, stir at room temperature for 12 hours, then use ethyl acetate The ester was extracted three times, and the organic phases were combined, washed once with saturated brine, dried, concentrated, dissolved in tetrahydrofuran (20 mL), added potassium tert-butoxide (1.24 g, 11.02 mmol) and stirred at room temperature for 3 hours, then added saturated aqueous ammonium chloride, ethyl acetate The ester was extracted three times, and the combined organic phases were washed with saturated brine, dried, concentrated, and purified on a silica gel column to obtain Compound 2 with a yield of 41%;
[0023] 1 H NMR (400MHz, CDCl 3)δ: 5.63-5.72 (m, 2H), 5.13-5.22 (m, 2H), 3.60-3.67 (ddd, J=10.0, 6.4, 3.2Hz, 1H), 3.18-3.25 (m, 1H)...
Embodiment 2
[0038] Step 1: Synthesis of (5S,6R)-6-allyl-5-(tert-butyldimethylsilyloxy)piperidin-2-one 2(P 1 =TBS)
[0039] Compound 1 (2.0 g, 5.51 mmol) was dissolved in tetrahydrofuran (50 mL), indium powder (1.27 g, 11.02 mmol) was added, allyl bromide (1.33 g, 11.02 mmol) was added dropwise after stirring. Stir at room temperature for 12 hours, concentrate, dilute with water, extract three times with ethyl acetate, combine the organic phases, wash once with saturated brine, dry, concentrate, dissolve in tetrahydrofuran (50 mL), and add lithium diisopropylamide after cooling down to -78 degrees (1.24g, 11.02mmol) After stirring for 3 hours at -78 degrees, add saturated aqueous ammonium chloride solution, extract three times with ethyl acetate, combine the organic phases with saturated brine, wash and dry, concentrate, and purify on a silica gel column to obtain compound 2. The yield is 46%;
[0040] 1 H NMR (400MHz, CDCl 3 )δ: 5.63-5.72 (m, 2H), 5.13-5.22 (m, 2H), 3.60-3.67 (ddd, J=...
Embodiment 3
[0044] Step 1: Synthesis of (5S,6R)-6-allyl-5-(tert-butyldimethylsilyloxy)piperidin-2-one 2(P 1 =TBS)
[0045] Compound 1 (2.0g, 5.51mmol) was dissolved in saturated sodium bromide aqueous solution (50mL), indium powder (1.27g, 11.02mmol) was added, allyl bromide (1.33g, 11.02mmol) was added dropwise after stirring, and room temperature After stirring for 12 hours, it was extracted three times with ethyl acetate, the organic phases were combined, washed once with saturated brine, dried and concentrated, dissolved in tetrahydrofuran (50mL), and added lithium diisopropylamide (1.24g, 11.02mmol ) after stirring at -78°C for 3 hours, adding saturated aqueous ammonium chloride solution, extracting with ethyl acetate three times, combining the organic phases, washing with saturated brine, drying, concentrating, and purifying on a silica gel column to obtain Compound 2 with a yield of 48%;
[0046] 1 H NMR (400MHz, CDCl 3 )δ: 5.63-5.72 (m, 2H), 5.13-5.22 (m, 2H), 3.60-3.67 (ddd, J...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More