

Process for stereoselective preparation of 4-bma using a chiral auxiliary
A technology of selection and solvent, which is applied in the field of stereoselective preparation of 4-BMA using chiral auxiliaries
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
Embodiment 1
[0063] Embodiment 1: Preparation of (S)-4-isopropyloxazolidin (isopropyloxazolidin)-2-ketone (2)
[0064]
[0065] The raw material L-valinol (150 g) was added to diethyl carbonate (227 ml), and then potassium carbonate (20 g) was added, while stirring the mixture at room temperature. The reaction solution was refluxed at 120-130° C. for 5 hours. The reaction solution was cooled to 0°C, 1.5N hydrochloric acid (450 ml) and ethyl acetate (450 ml) were added, and the resulting two phases were separated. The aqueous phase was extracted twice with ethyl acetate (450ml), the organic phase was washed with aqueous sodium chloride (450ml), the phases were separated, dried, filtered and distilled. Diisopropyl ether (225 ml) was added to produce crystals, to which n-hexane (225 ml) was added. The mixture was stirred at 0 °C for 1 h, then filtered and dried to give the title compound (170 g, 85% yield).
[0066] 1 H NMR (300MHz, CDCl 3 )δ4.4(t, 1H), 4.1(m, 1H), 3.6(q, 1H), 1.7(m, ...
Embodiment 2
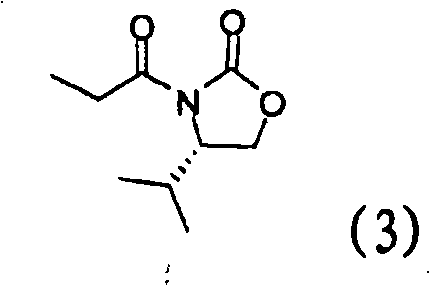
[0067] Example 2: Preparation of (S)-4-isopropyl-3-propionyl oxazolidin-2-one (3)
[0068]
[0069] Compound (2) (100 g) prepared in Example 1 was dissolved in tetrahydrofuran (300 ml), and cooled to 0°C. Lithium chloride (36 g) was added followed by triethylamine (101 g) slowly and the resulting mixture was stirred for 30 min. Propionic anhydride (106 g) was slowly added over a period of 30 min. The reaction mixture was slowly warmed to room temperature and stirred for 1-1.5 h. The reaction solution was cooled, 1N aqueous sodium chloride solution (300 ml) was added, and the mixture was stirred for 30 min. Ethyl acetate (300ml) was added, the phases were separated and extracted once more with ethyl acetate (300ml). After washing with 1.5N hydrochloric acid (300ml), the organic phase was washed once more with aqueous sodium chloride (300ml), dried, filtered and distilled to give the title compound (142g, 99% yield).
[0070] 1 H NMR (300MHz, CDCl 3 )δ4.4(m, 1H), 4.3-4....
Embodiment 3
[0071] Example 3: (S)-3-((R)-2-(3-((R)-1-(tert-butyldimethylsilyloxy)ethyl)-4-oxoazepine Preparation of cyclobutan-2-yl)propionyl)-4-isopropyloxazolidin-2-one (5)
[0072]
[0073] Compound (3) (44 g) prepared in Example 2 was dissolved in dichloromethane (890 ml), and cooled to 0°C. Titanium chloride (55 g) was added slowly. After 1 h, diisopropylethylamine (40 g) was added followed by 4-AA (50 g). The resulting mixture was reacted at room temperature for 3 h and cooled. Water (890ml) was added to separate the phases, and 1.5N hydrochloric acid (500ml) was added thereto. The phases were separated and washed once more with aqueous sodium bicarbonate, with aqueous sodium chloride (100ml), dried over magnesium sulfate and distillation to yield the title compound (95g) contaminated with some impurities.
[0074] 1 H NMR (300MHz, CDCl 3 )δ5.96(s, 1H), 4.44(m, 1H), 4.30(m, 4H), 3.96(m, 1H), 3.05(m, 1H), 2.30(m, 1H), 1.25(dd, 6H ), 0.92(m, 15H), 0.07(d, 6H)
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More