

High-throughput sequencing data-based genome de novo assembly method
A sequencing data and assembly method technology, applied in the direction of electrical digital data processing, special data processing applications, instruments, etc., can solve the problems of high proportion of repeated sequences, reduced assembly effect, and increased assembly difficulty
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

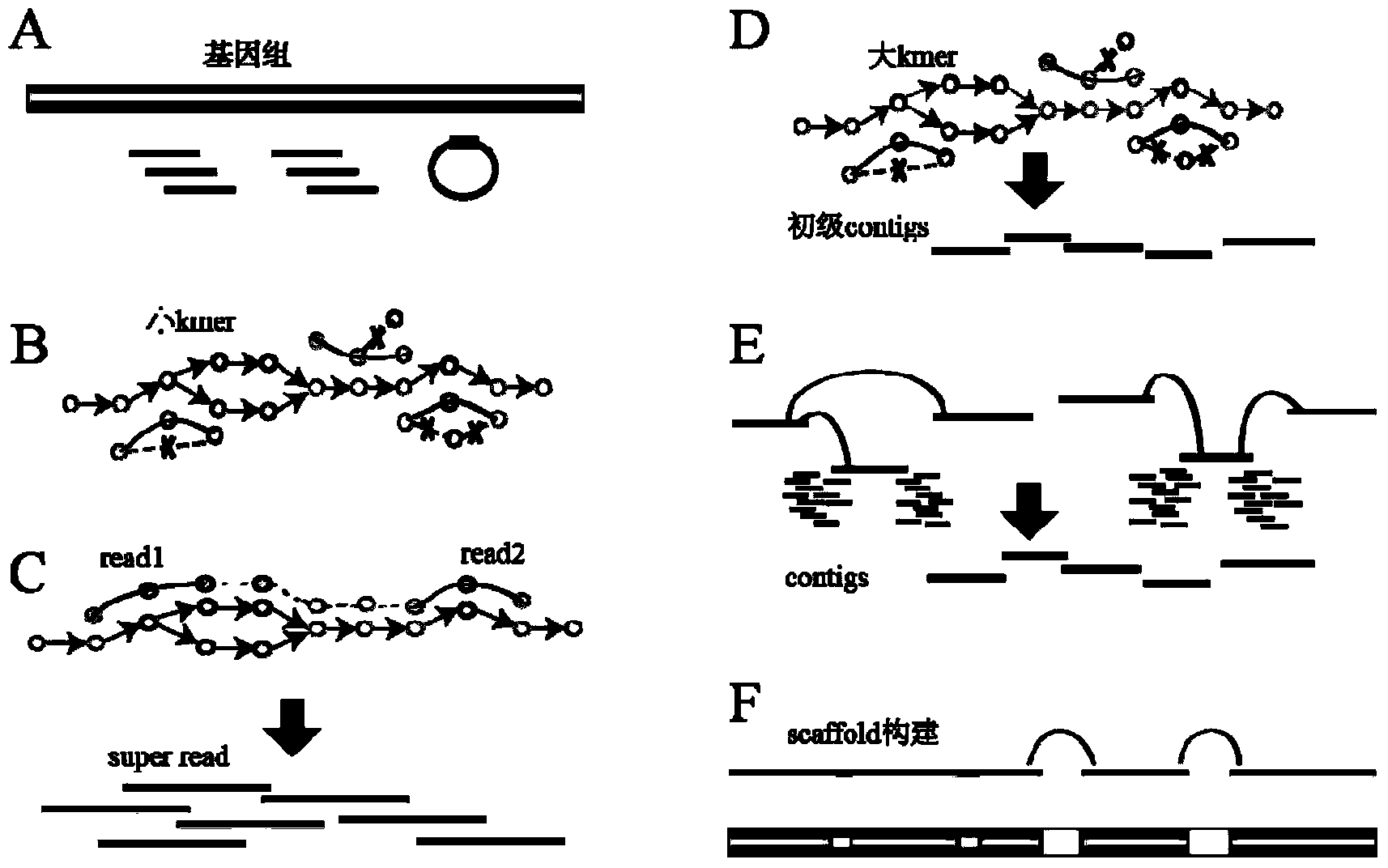
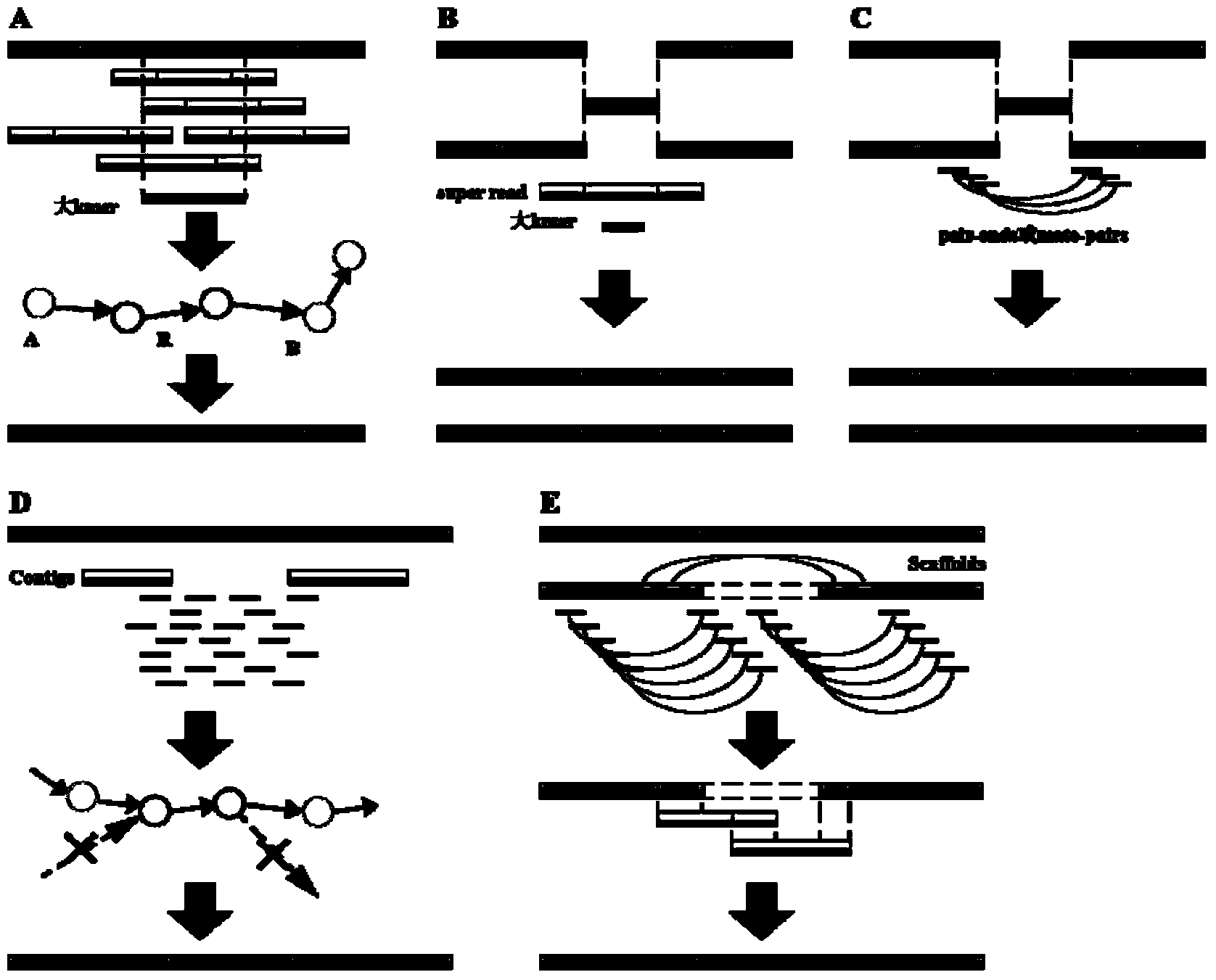
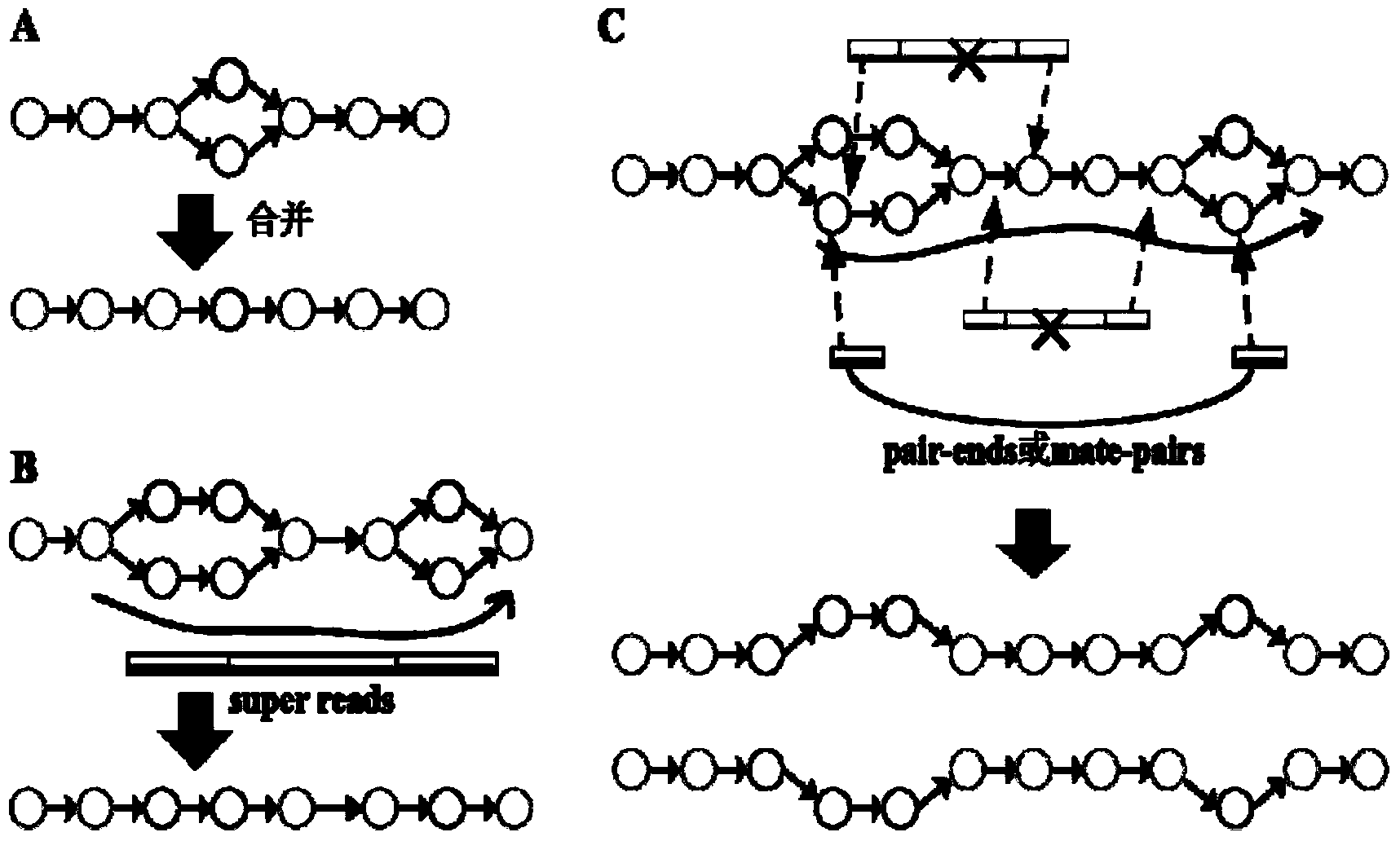
Examples
Embodiment 1
[0064] Example 1 Escherichia coli (E.coli) genome assembly
[0065] 1) Test data introduction
[0066] The test data is downloaded from the SRA (Short Read Archive) database of NCBI (National Center for Biotechnology Information, namely the National Center for Biotechnology Information), the SRA database website is www.ncbi.nlm.nih.gov / sra, the data The detailed accession number is SRX016044. The details of the test data are as follows:
[0067] Upload date: 2009-05-22;
[0068] Library size: 180bp;
[0069] Total sequencing volume: 2.1G;
[0070] Predicted genome sequencing depth: 456.5x.
[0071] 2) Evaluation method
[0072] A total of 7 assembly software were tested and compared, the main parameters of each assembly software were traversed, and then the result with the best assembly result was selected for comparison and evaluation. The detailed assembly parameters of the best assembly results of each software are as follows:
[0073] GNOVO (the inventive method) as...
Embodiment 2
[0105] Example 2 Streptomyces (S.roseosporus) genome assembly
[0106] 1) Test data introduction
[0107] The test data is downloaded from NCBI's SRA database, the website of the SRA database is www.ncbi.nlm.nih.gov / sra, and the detailed accession numbers of the data are SRX026747 and SRX016085.
[0108] a) The details of the test data SRX026747 are as follows:
[0109] Upload date: 2010-08-06;
[0110] Library size: 180bp;
[0111] Total sequencing volume: 10.7G;
[0112] Predicted genome sequencing depth: 1389.6X.
[0113] b) The details of the test data SRX016085 are as follows:
[0114] Upload date: 2009-09-20;
[0115] Library size: 4kb;
[0116] Total sequencing volume: 3.5G;
[0117] Predicted genome sequencing depth: 454.5X.
[0118] 2) Evaluation method
[0119] Here, a total of 5 assembly software are tested and compared, the main parameters of each assembly software are traversed, and then the result with the best assembly result is selected for comparison...
Embodiment 3
[0131] Example 3 Neurospora crassa (N.crassa) genome assembly
[0132] 1) Test data introduction
[0133] The test data is downloaded from NCBI's SRA database. The website of the SRA database is www.ncbi.nlm.nih.gov / sra, and the detailed accession number of the data is SRX030834.
[0134] a) The details of the test data SRX030834 are as follows:
[0135] Upload date: 2010-11-11;
[0136] Library size: 180bp;
[0137] Total sequencing volume: 5.5G;
[0138] Predicted genome sequencing depth: 148.3X.
[0139] 2) Evaluation method
[0140] A total of 6 assembly software are tested and compared here. Here, the main parameters of each assembly software are traversed, and then the result with the best assembly result is selected for comparison and evaluation. The detailed assembly parameters of the best assembly results of each software are as follows:
[0141] GNOVO assembly parameters are: k1=25, k2=95, m1=5, m2=2, other parameters are default parameters (detailed evaluation...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More