Cytosine methylation excavation method
A technology of cytosine methylation and methylation-sensitive enzymes, applied in the field of bioinformatics, can solve the problems of cumbersome process, time-consuming, expensive and expensive, and achieve the effect of accelerating research and development and low cost
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
Embodiment Construction
[0022] The present invention will be described in further detail below in conjunction with the accompanying drawings and specific embodiments.
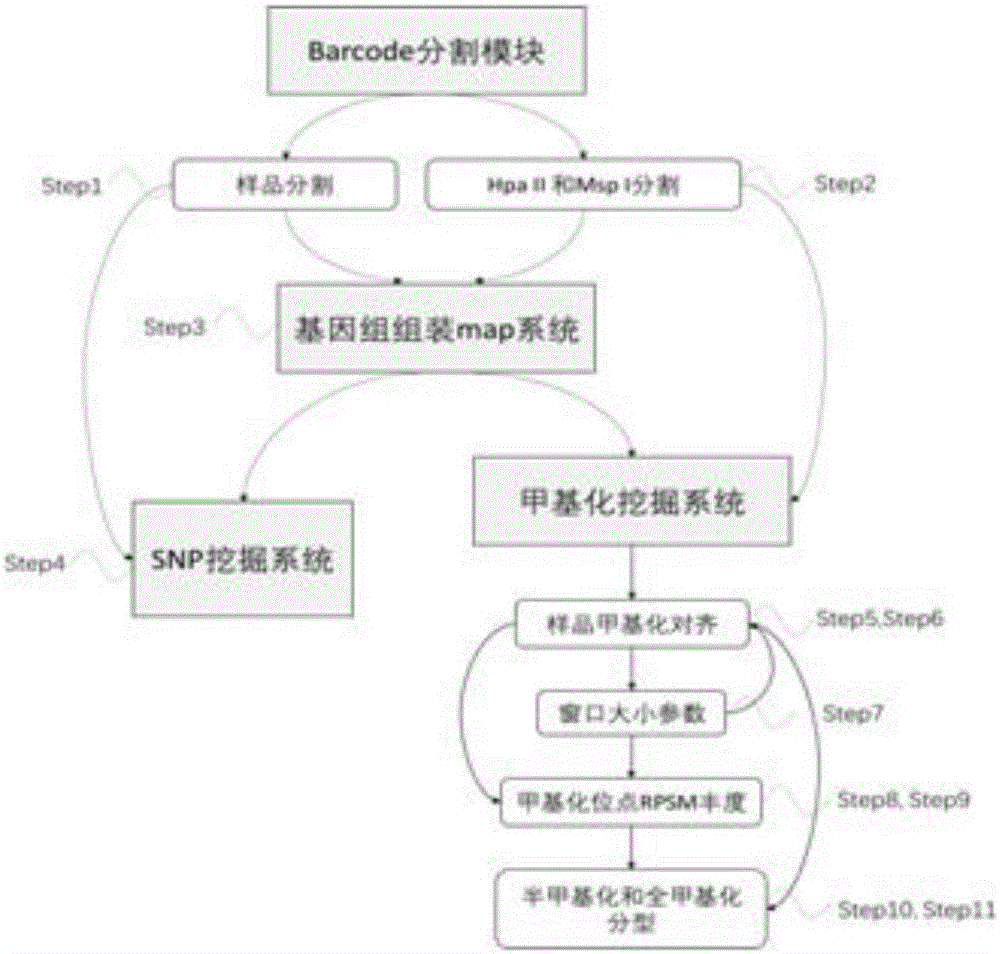
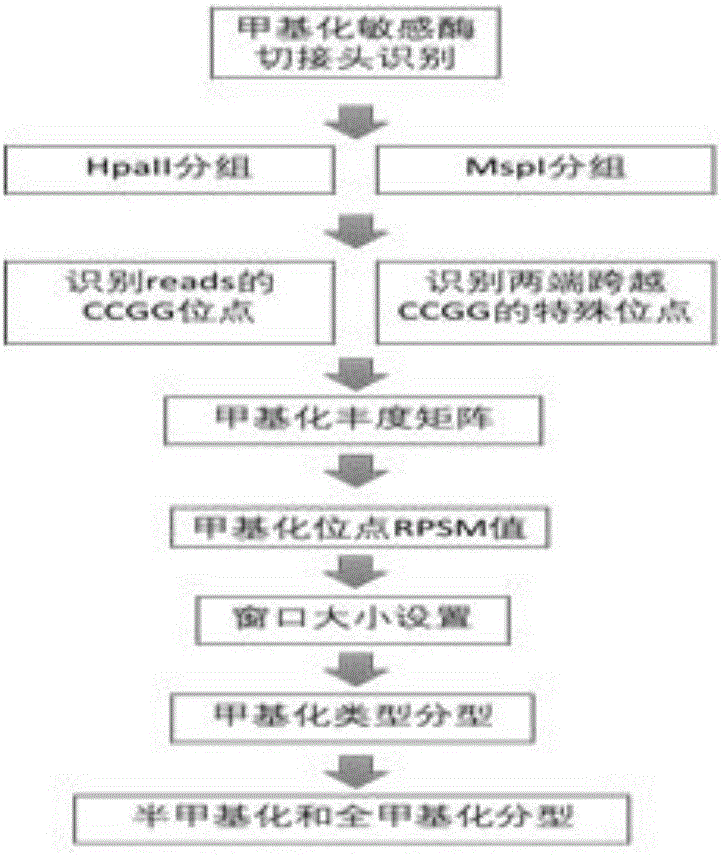
[0023] The main steps of the process of the methylation typing software of the present invention are as follows: figure 1 As shown, the core module of methylation typing is as follows figure 2 Shown: Step1, source high-throughput sequencing raw reads, use the barcode segmentation module to divide the reads into multiple sample reads according to the barcode.
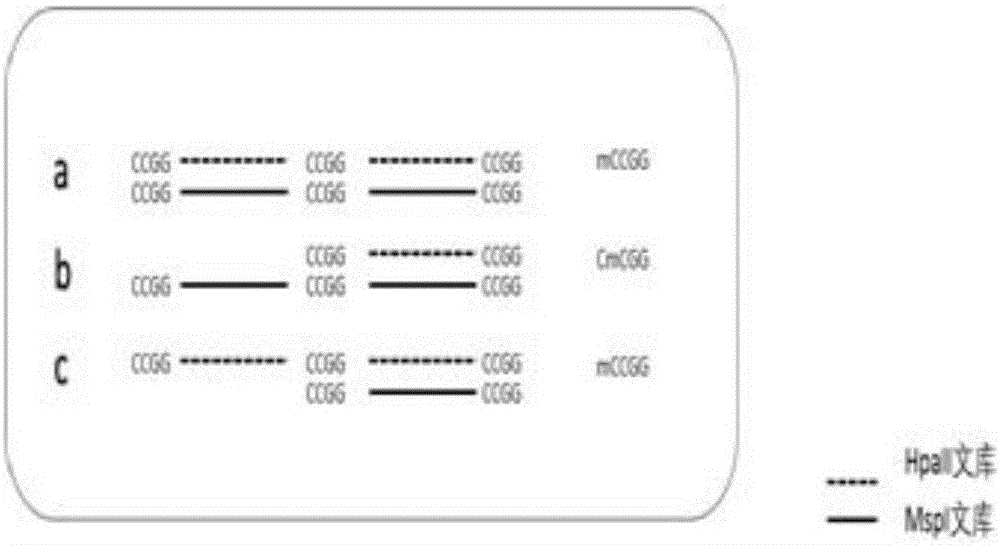
[0024] Step2. Use the barcode segmentation module to divide the read into two methylation pools, HpaII and MspI;
[0025] Step3. Mix all the barcode-processed reads, and map the reads to the reference genome based on bowtie2 to perform the assembly step.
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More