

Novel synthesis method of Selinexor active pharmaceutical ingredient
A synthesis method and a technology for an API, which are applied in the new synthesis field of Selinexor API, can solve the problems of affecting the yield and being difficult to remove, and achieve the effect of reducing the synthesis steps and improving the yield.
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

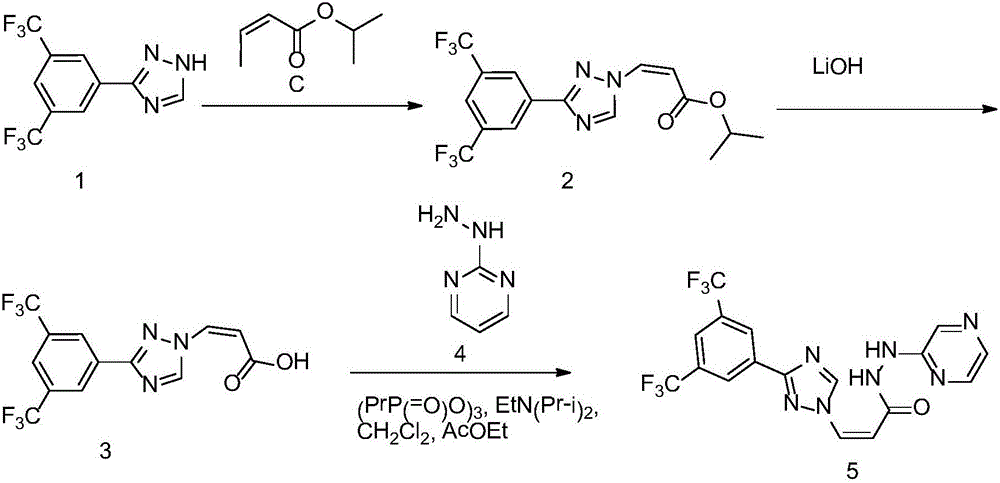
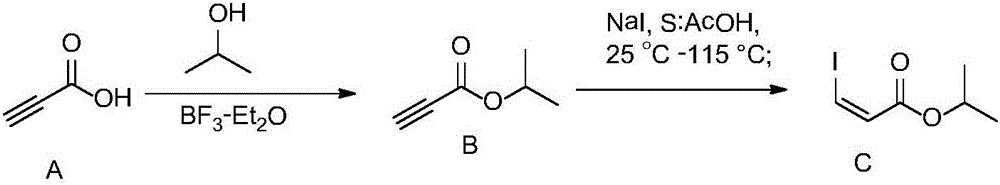
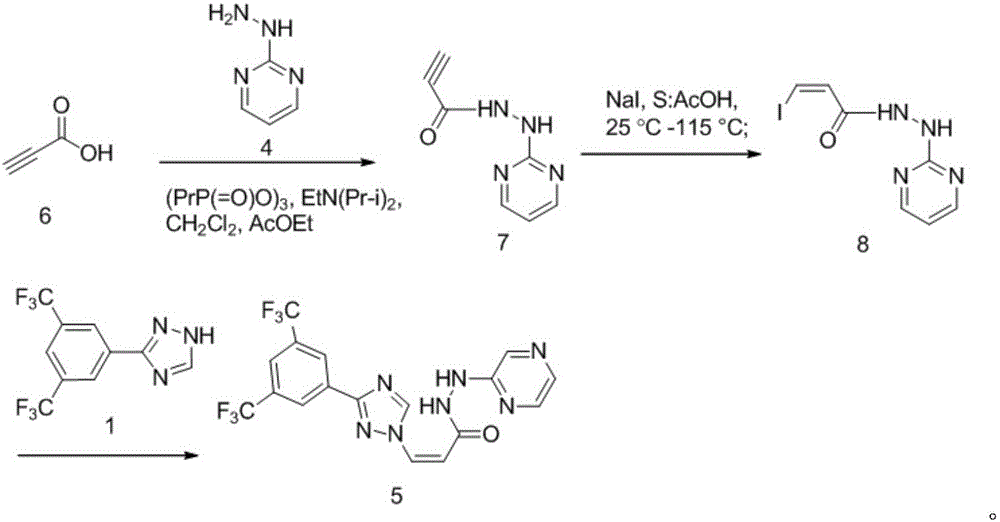
Examples
Embodiment 1
[0024] A novel synthetic method of Selinexor bulk drug, comprising the following steps:
[0025] A, the synthesis of compound 7
[0026] In a 50ml three-neck flask, add 0.2g of compound 6, 15ml of dichloromethane and 15ml of ethyl acetate, stir and dissolve, then add 0.3g of compound 4 and 3g of T 3 P, 0.75g DIPEA; the system was stirred at 0°C for 30 minutes to react, after the reaction was completed, 50ml of dichloromethane and 30ml of water were added, the liquid was separated, and the organic phase was evaporated to dryness to obtain the crude compound 7, which was directly cast down without purification;
[0027] B, the synthesis of compound 8
[0028] Add compound 7, 40ml glacial acetic acid and 1.38g sodium iodide obtained in the previous step to a 50ml three-necked flask, raise the temperature to 115°C, and react for 3h; Water and 100ml of dichloromethane were stirred for 10min, and the layers were allowed to stand. The organic phase was washed with saturated sodium ...
Embodiment 2
[0032] A novel synthetic method of Selinexor bulk drug, comprising the following steps:
[0033] A, the synthesis of compound 7
[0034] In a 50ml three-neck flask, add 0.2g of compound 6, 15ml of dichloromethane and 15ml of ethyl acetate, stir and dissolve, then add 0.3g of compound 4 and 3g of T 3 P, 0.75g DIPEA; the system was stirred at 1°C for 35 minutes to react, after the reaction was completed, 50ml of dichloromethane and 30ml of water were added, the liquid was separated, and the organic phase was evaporated to dryness to obtain the crude compound 7, which was directly cast down without purification;
[0035] B, the synthesis of compound 8
[0036] Add compound 7, 40ml glacial acetic acid and 1.38g sodium iodide obtained in the previous step to a 50ml three-necked flask, raise the temperature to 120°C, and react for 2.5h; 60ml of water and 120ml of dichloromethane, after stirring for 15min, let stand to separate layers, the organic phase was washed with saturated so...
Embodiment 3
[0040] A novel synthetic method of Selinexor bulk drug, comprising the following steps:
[0041] A, the synthesis of compound 7
[0042] In a 50ml three-necked flask, add 0.2g of compound 6, 15ml of dichloromethane and 15ml of ethyl acetate, stir and dissolve, then add 0.3g of compound 4 and 3g of T 3 P, 0.75g DIPEA; the system was stirred at 0°C for 25 minutes to react, after the reaction was completed, 40ml of dichloromethane and 35ml of water were added, the liquid was separated, and the organic phase was evaporated to dryness to obtain the crude compound 7, which was directly cast down without purification;
[0043] B, the synthesis of compound 8
[0044] Add compound 7, 35ml glacial acetic acid and 1.38g sodium iodide obtained in the previous step to a 50ml three-necked flask, raise the temperature to 110°C, and react for 3.5h; 50ml of water and 100ml of dichloromethane were stirred for 8min, and the layers were separated. The organic phase was washed with saturated sod...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More