

Improved tofacitinib synthesis method and impurity preparation method
A synthetic method, the technology of tofacitinib, which is applied in the field of medicinal chemistry, can solve the problems of tofacitinib degradation and other problems, and achieve the effect of easy removal and quality improvement
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
Embodiment 1
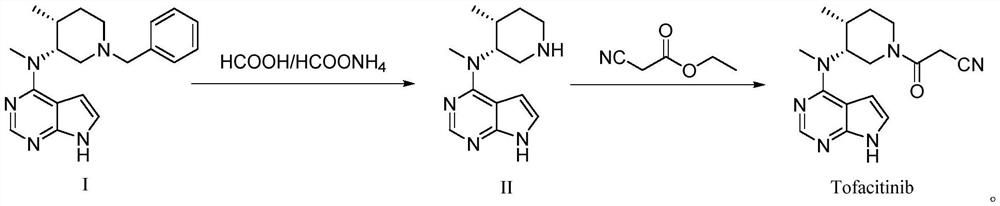
[0035] Add 6.0 g of Compound I, 1.2 g of 10% Pd / C, 48.0 g of ammonium formate, and 72 ml of methanol into the reaction flask, heat to 65° C., and keep stirring for 9 h. After the reaction was complete, it was filtered, extracted with dichloromethane and ethyl acetate, washed with water and distilled under reduced pressure to obtain 3.0 g of white powder Compound II with a yield of 68.2%.
[0036] Add 3.0 g of Compound II, 1.4 g of ethyl cyanoacetate, 1.2 g of DBU, and 10 ml of n-butanol into the reaction flask, raise the temperature to 60-65°C, and keep stirring for 8-9 hours. After the reaction was complete, it was lowered to room temperature, added with water, extracted with ethyl acetate, concentrated and dried to obtain 1.6 g of light yellow tofacitinib crude product, with a yield of 62.8%.
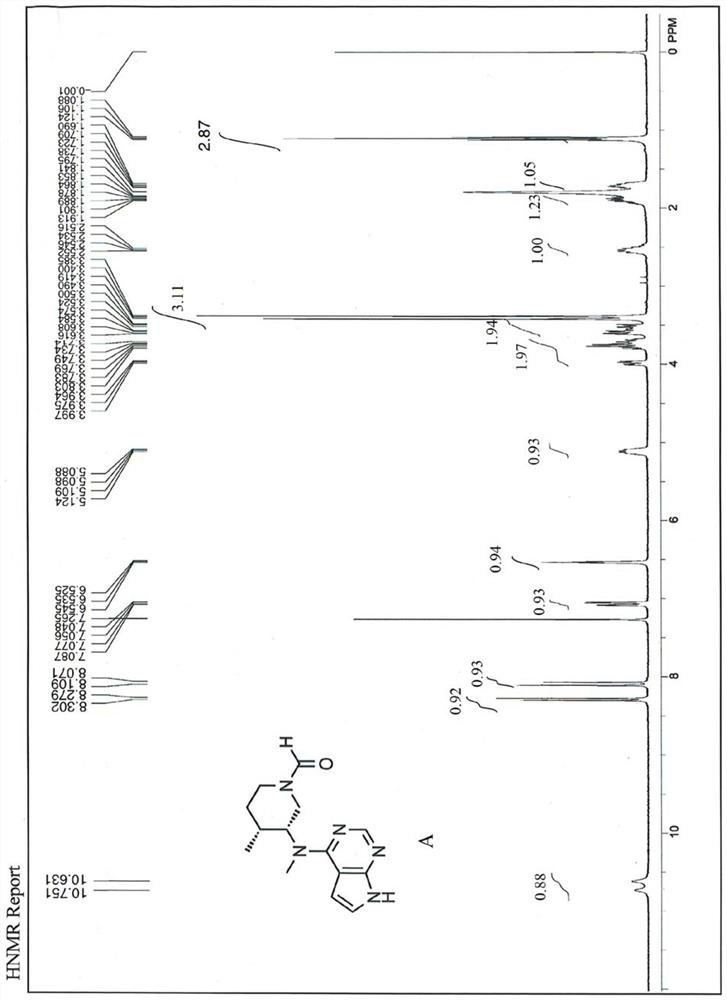
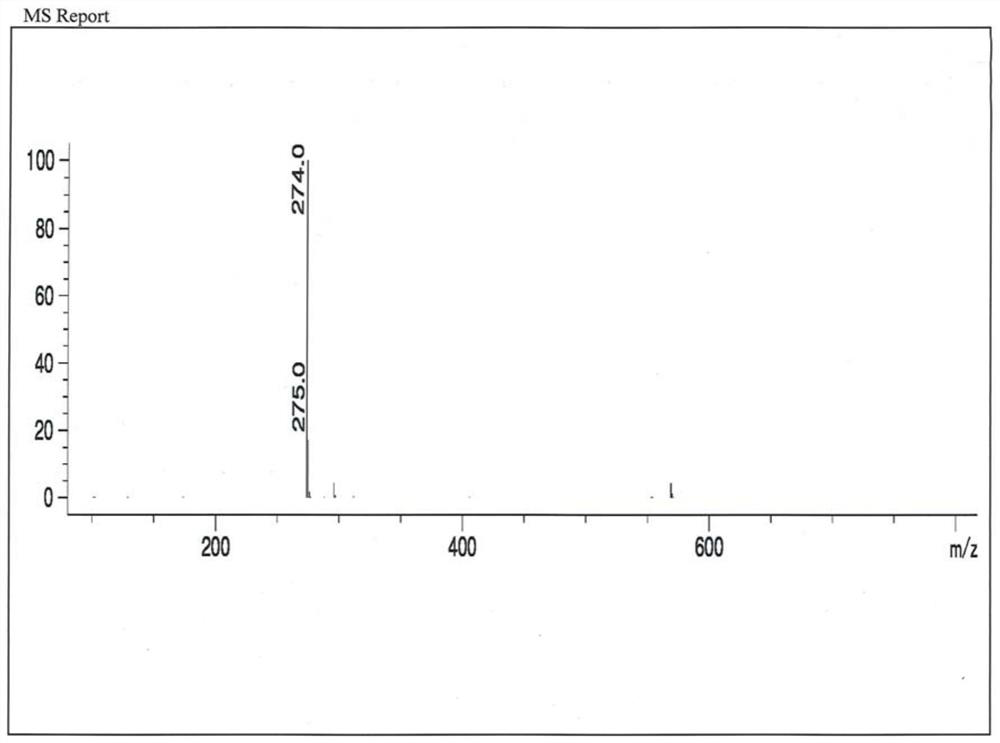
[0037] The impurity formula A was separated from the compound II by chromatographic separation, which accounted for 32.6% by weight of the product. As the reaction time increases, th...
Embodiment 2
[0039] Add 40 g of Compound I, 8 g of 10% Pd / C, 233 g of formic acid, and 480 ml of methanol into the reaction flask, heat to 67-70°C, and keep stirring for 4-5 hours. After the reaction was complete, it was cooled to room temperature and filtered. After the filtrate was concentrated under reduced pressure, the pH was adjusted to 14 by adding 30% aqueous sodium hydroxide solution, and the aqueous phase was washed with ethyl acetate. Then methanol was added to the water phase, and the reaction was stirred at 50-60°C for 4h. After the reaction was complete, dichloromethane was added for extraction three times, the liquid was separated, the organic phase was concentrated until no distillate was present, and then dried to obtain 24.1 g of compound II as a white solid powder with a yield of 82.4%. Impurity formula A was not detected.
[0040] Add 20g of compound II, 18.4g of ethyl cyanoacetate, 12.4g of DBU, and 100ml of tetrahydrofuran into the reaction flask, heat up to 60-65°C,...
Embodiment 3
[0042] Add 40 g of compound I, 8 g of 10% Pd / C, 320 g of ammonium formate, and 480 ml of methanol into the reaction flask, heat to 67-70°C, and keep stirring for 4-5 hours. After the reaction was complete, it was cooled to room temperature and filtered. After the filtrate was concentrated under reduced pressure, the pH was adjusted to 13 by adding 30% aqueous sodium hydroxide solution, and the aqueous phase was washed with ethyl acetate. Then methanol was added to the water phase, and the reaction was stirred at 55-60°C for 4h. After the reaction was complete, dichloromethane was added for extraction three times, the liquid was separated, the organic phase was concentrated until no distillate was present, and then dried to obtain 22.3 g of compound II as a white solid powder. Yield 76.2%. Impurity formula A was not detected.
[0043] Add 20g of compound II, 18.4g of ethyl cyanoacetate, 12.4g of DBU, and 100ml of tetrahydrofuran into the reaction flask, heat up to 60-65°C, an...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More