[0029] In a further aspect of the invention, a method for identifying nucleic acid sequences that encode at least two interacting proteins is provided, comprising the steps of a) combining (i) a first vector comprising (1) a nucleic acid sequence that encodes a first protein that interacts with a second protein, and (2) a first restriction endonuclease recognition site specific for a restriction endonuclease that cleaves the nucleic acid sequence fragment distally to the restriction endonuclease recognition site, and (ii) a second vector comprising (1) a nucleic acid sequence that encodes the second protein; and (2) a second restriction endonuclease recognition site specific for a restriction endonuclease that cleaves the nucleic acid sequence fragment distally to the restriction endonuclease recognition site, thereby producing a combination, b) optionally maintaining the combination under conditions in which the first protein and the second protein are expressed and interact, c) joining the first vector with the second vector, thereby forming a contiguous nucleic acid sequence that comprises (i) the nucleic acid sequence that encodes a first protein that interacts with a second protein, (ii) the first restriction endonuclease recognition site specific for a restriction endonuclease that cleaves the nucleic acid sequence fragment distally to the restriction endonuclease recognition site, (iii) the nucleic acid sequence that encodes the second protein, and (iv) the second restriction endonuclease recognition site specific for a restriction endonuclease that cleaves the nucleic acid sequence fragment distally to the restriction endonuclease recognition site, d) cleaving the first restriction endonuclease recognition site and the second restriction endonuclease recognition site in the contiguous nucleic acid sequence with restriction endonucleases that cleave the contiguous nucleic acid sequence distally to the restriction endonuclease recognition sites, thereby producing a 5′ end tag and a 3′ end tag of the contiguous nucleic acid sequence, e) joining the 5′ end tag to the 3′ end tag, thereby producing a paired tag, and f) sequencing the paired tag, thereby identifying nucleic acid sequences that encode at least two interacting proteins.
[0030] In an additional embodiment of the invention, provided is a method for identifying nucleic acid sequences that encode at least two interacting proteins comprising the steps of a) combining (i) a first vector comprising (1) a nucleic acid sequence that encodes a first protein that interacts with a second protein, and (2) a first restriction endonuclease recognition site specific for a restriction endonuclease that cleaves the nucleic acid sequence fragment distally to the restriction endonuclease recognition site, and (ii) a second vector comprising (1) a nucleic acid sequence that encodes the second protein, and (2) a second restriction endonuclease recognition site specific for a restriction endonuclease that cleaves the nucleic acid sequence fragment distally to the restriction endonuclease recognition site, thereby producing a combination, b) optionally maintaining the combination under conditions in which the first protein and the second protein are expressed and interact, c) joining the first vector with the second vector, thereby forming a contiguous nucleic acid sequence that comprises (i) the nucleic acid sequence that encodes a first protein that interacts with a second protein, (ii) the first restriction endonuclease recognition site specific for a restriction endonuclease that cleaves the nucleic acid sequence fragment distally to the restriction endonuclease recognition site, (iii) the nucleic acid sequence that encodes the second protein, and (iv) the second restriction endonuclease recognition site specific for a restriction endonuclease that cleaves the nucleic acid sequence fragment distally to the restriction endonuclease recognition site, d) sequencing the contiguous nucleic acid sequence, thereby identifying nucleic acid sequences that encode at least two interacting proteins.
[0031] In another embodiment of the invention, provided is a method for identifying nucleic acid sequences that encode at least two interacting proteins comprising the steps of a) combining, (i) a first vector comprising (1) a nucleic acid sequence that encodes a first protein that interacts with a second protein, and (2) a first restriction endonuclease recognition site specific for a restriction endonuclease that cleaves the nucleic acid sequence fragment distally to the restriction endonuclease recognition site, and (ii) second vector comprising (1) a nucleic acid sequence that encodes the second protein; and (2) a second restriction endonuclease recognition site specific for a restriction endonuclease that cleaves the nucleic acid sequence fragment distally to the restriction endonuclease recognition site, thereby producing a combination, b) optionally maintaining the combination under conditions in which the first protein and the second protein are expressed and interact, c) joining the first vector with the second vector, thereby forming a contiguous nucleic acid sequence that comprises (i) the nucleic acid sequence that encodes a first protein that interacts with a second protein, (ii) the first restriction endonuclease recognition site specific for a restriction endonuclease that cleaves the nucleic acid sequence fragment distally to the restriction endonuclease recognition site, (iii) the nucleic acid sequence that encodes the second protein, and (iv) the second restriction endonuclease recognition site specific for a restriction endonuclease that cleaves the nucleic acid sequence fragment distally to the restriction endonuclease recognition site, d) cleaving the first restriction endonuclease recognition site and the second restriction endonuclease recognition site in the contiguous nucleic acid sequence with restriction endonucleases that cleave the contiguous nucleic acid sequence distally to the restriction endonuclease recognition sites, thereby producing a paired tag comprising a 5′ end tag and a 3′ end tag of the contiguous nucleic acid sequence, e) sequencing the paired tag, thereby identifying nucleic acid sequences that encode at least two interacting proteins.
[0032] In a further embodiment of the invention, provided is a method for identifying a plurality of nucleic acid sequences that encode at least two interacting proteins comprising the steps of a) combining (i) a plurality of first vectors each comprising (1) a nucleic acid sequence that encodes a first protein that interact with a second protein; and (2) a first restriction endonuclease recognition site specific for a restriction endonuclease that cleaves the nucleic acid sequence fragment distally to the restriction endonuclease recognition site, and (ii) a plurality of second vectors each comprising (1) a nucleic acid sequence that encodes the second protein; and (2) a second restriction endonuclease recognition site specific for a restriction endonuclease that cleaves the nucleic acid sequence fragment distally to the restriction endonuclease recognition site, thereby producing a combination comprising a plurality of first vectors and a plurality of second vectors, b) optionally maintaining the combination under conditions in which the plurality of first vectors encoding a first protein and the p



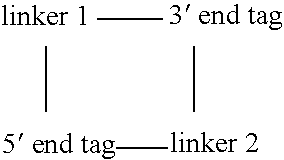
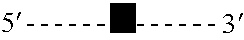
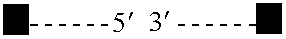
 Login to View More
Login to View More