

Method for synthesizing intermediate of cefcapene pivoxil,
A technology of cefcapene pivoxil and synthetic method, which is applied in the direction of organic chemistry, etc., to achieve the effects of simple operation, reduced preparation cost, and reduced reaction steps
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
reference example 1
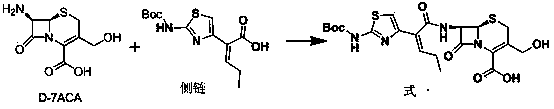
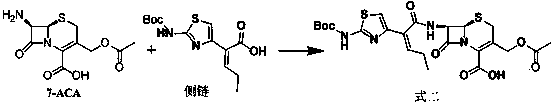
[0033] Reference Example 1 7β-[(Z)-2-(2-tert-butoxycarbonylaminothiazol-4-yl)-2-butenoyl]amino-3-acetoxymethyl-3-cephem-4-carboxy Synthesis of acid (Formula 2) (refer to US4500716)
[0034] Add 142g (0.5mol) of (Z)-2-(2-tert-butoxycarbonylaminothiazol-4-yl)-2-butenoic acid and 76mL (0.55mol) of triethylamine into dichloromethane (400mL) and cool down To -50°C, 40mL (0.52mol) of methanesulfonyl chloride was added dropwise, the temperature was controlled between -50°C to -40°C, and the drop was completed, and stirred at -50°C to -40°C for 5h. Then, add 163g (0.6mol) of 7-ACA (0.6mol) and 180mL (1.3mol) of triethylamine in dichloromethane (400mL) dropwise to the reaction solution, and control the temperature between -50°C and -40°C. ℃~-40℃ for 3h. Acidify with dilute hydrochloric acid and extract with ethyl acetate. The extract was washed with brine and dilute sodium bicarbonate water, dried and concentrated to give 7β-[(Z)-2-(2-tert-butoxycarbonylaminothiazol-4-yl)-2-butenoyl...
Embodiment 1
[0035] Example 1 7β-[(Z)-2-(2-tert-butoxycarbonylaminothiazol-4-yl)-2-butenoyl]amino-3-hydroxymethyl-3-cephem-4-carboxylic acid (formula 1) synthesis
[0036] Add 20g (36.2mmol) of compound formula 2 into 100mL of purified water, lower the temperature to -15°C, adjust the pH to 8 with sodium hydroxide, and keep the reaction for 3 hours. After the reaction is completed, add 10g of tetra-n-butylammonium bisulfate, dichloromethane (50mL×2) extraction, take the dichloromethane layer, concentrate the dichloromethane to cut off the flow, add isopropyl ether to the residue, stir and crystallize, filter with suction, and dry to obtain compound formula 1 18.0g, yield 98%, content 94 %.
Embodiment 2
[0037] Example 2 7β-[(Z)-2-(2-tert-butoxycarbonylaminothiazol-4-yl)-2-butenoyl]amino-3-hydroxymethyl-3-cephem-4-carboxylic acid (formula 1) synthesis
[0038] Add 20g (36.2mmol) of compound formula 2 into 50mL of acetone and 100mL of water, cool down to -30°C, adjust the pH to 7 with sodium carbonate, and keep the reaction for 10h. After the reaction is completed, add 10g of tetra-n-butylammonium chloride, Extracted with methyl chloride (50mL×2), took the dichloromethane layer, concentrated the dichloromethane until the flow was cut off, added isopropyl ether to the residue, stirred and crystallized, filtered with suction, and dried to obtain 17.8g of compound formula 1, with a yield of 96%. Content 90%.
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More