

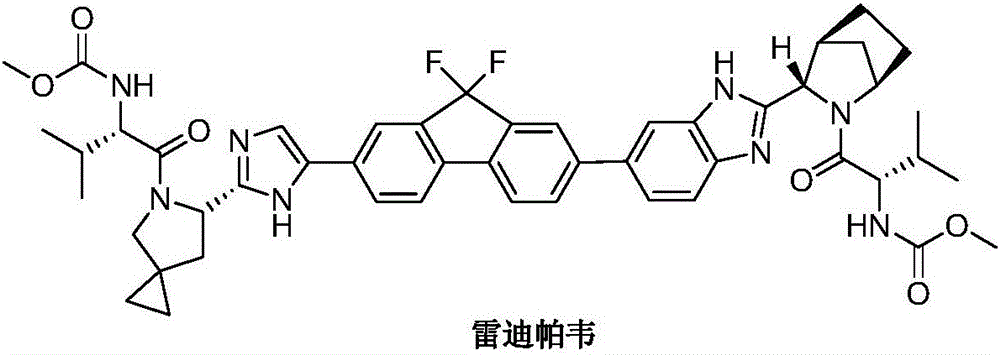
Preparation method of ledipasvir intermediate and intermediate compound
A technology for compounds and intermediates, applied in the field of preparation of intermediates, can solve the problem of high cost, achieve the effects of improving total yield, improving market competitiveness, and improving atomic economic efficiency
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

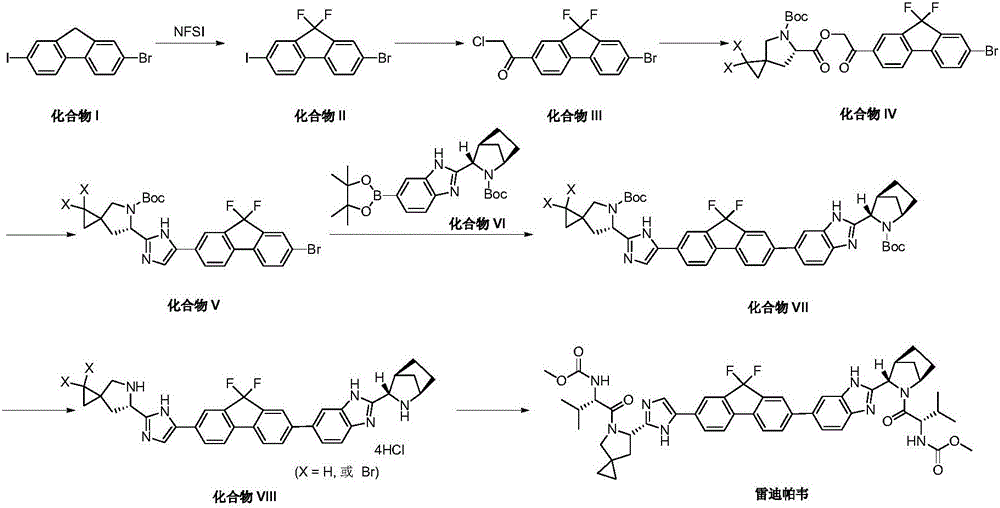
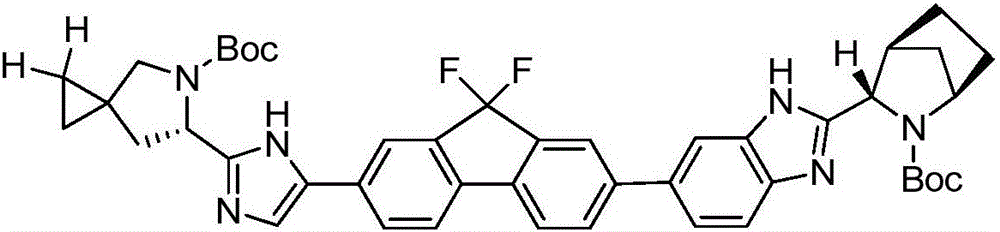
Examples
Embodiment 1
[0040] The synthesis of embodiment 1 compound XI
[0041] Add 100 g of compound X, 66 g of N-chlorosuccinimide and 200 mL of acetonitrile into a 1.0 L reaction flask. Stir to dissolve, cool down to -2°C, and add 44 mL of concentrated hydrochloric acid dropwise. At the end of the dropwise addition, the temperature was 5°C, and the temperature was naturally raised to 20±5°C, and the reaction was stirred for 4.5 hours. Sampling was sent to LC-MS detection to show that the reaction was complete. The reaction solution was filtered, and the filter cake was washed twice with 300 mL of methanol. Add 500 mL of n-heptane to the filter cake and stir for 1 hour. After filtering, the filter cake was washed with 200 mL of methanol, and the filter cake was dried to obtain 80.2 g of compound XI as a white solid (purity 91.37%, yield 71.7%). 1 H NMR (400MHz, CDCl 3 ), δ: 7.66(d, J=8.5Hz, 1H), 7.60(d, J=8.1Hz, 1H), 7.51(s, 1H), 7.36(d, J=8.0Hz, 1H), 3.87(s ,1H).
Embodiment 2
[0042] The synthesis of embodiment 2 compound XII
[0043]Add 60g of compound XI, 186g of N-fluorobisbenzenesulfonamide and 660mL of tetrahydrofuran into a 2.0L reaction flask. Under the protection of nitrogen, the reaction solution is cooled to -70°C, and 690mL of bis(trimethylsilyl)amino is slowly added dropwise. Lithium tetrahydrofuran solution, the temperature is controlled at -60~-70°C during the dropwise addition. After the dropwise addition, keep it warm at -60~-70°C, stir and react for 2 hours, take samples and send them to LC-MS for detection, which shows that the reaction is over, and the temperature rises naturally To -20°C, add 108mL of water dropwise until clear. Concentrate under reduced pressure until a large amount of solids are precipitated, add 840 mL of distilled tetrahydrofuran with water in three batches, and form a good solid. Cool to 20°C and filter. Add 180 g of methanol to the filter cake, heat up to 40°C and stir for 30 minutes, cool down to 20°C an...
Embodiment 3
[0045] The synthesis of embodiment 3 compound XIII
[0046] Add 30g compound XII, 29g biboronic acid pinacol ester, 19g potassium acetate, 6.94g Pd(dppf)Cl in 1.0L one-mouth bottle 2 and 500 mL of 1.4-dioxane. In a stirring state, nitrogen was replaced three times, and the temperature was raised to 70° C. under a nitrogen atmosphere and stirred for 2.5 hours. Sampling was sent to LC-MS to detect that the reaction was complete, and 250 mL of water was added to quench the reaction, and 500 mL of ethyl acetate was added for extraction, and the aqueous phase was extracted once with 500 mL of ethyl acetate. The organic phases were combined, washed twice with saturated brine, and concentrated to dryness under reduced pressure to obtain a crude product. The crude product was dissolved in 140 mL of methanol, stirred at 60° C. for 2 hours and filtered to obtain 28.4 g of compound XIII (yield 83%, HPLC purity 92%). 1 H NMR (400MHz, CDCl 3 )δ8.07(s,1H),7.92(d,J=7.5Hz,1H),7.60(d,J=1.1...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More