

Drug target affinity prediction method based on deep learning
A deep learning and prediction method technology, applied in drug reference, biological neural network model, neural architecture, etc., can solve the problem of affecting prediction accuracy, drug-target interaction accuracy is not high, ignoring drug-target interaction combination Affinity and other issues to achieve the effect of improving fault tolerance, compressing the number, and reducing overfitting
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
Embodiment 1
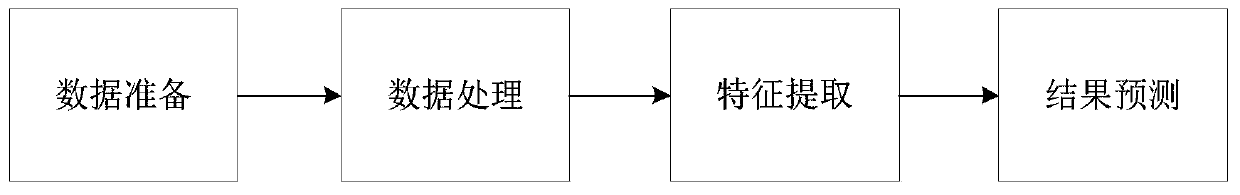
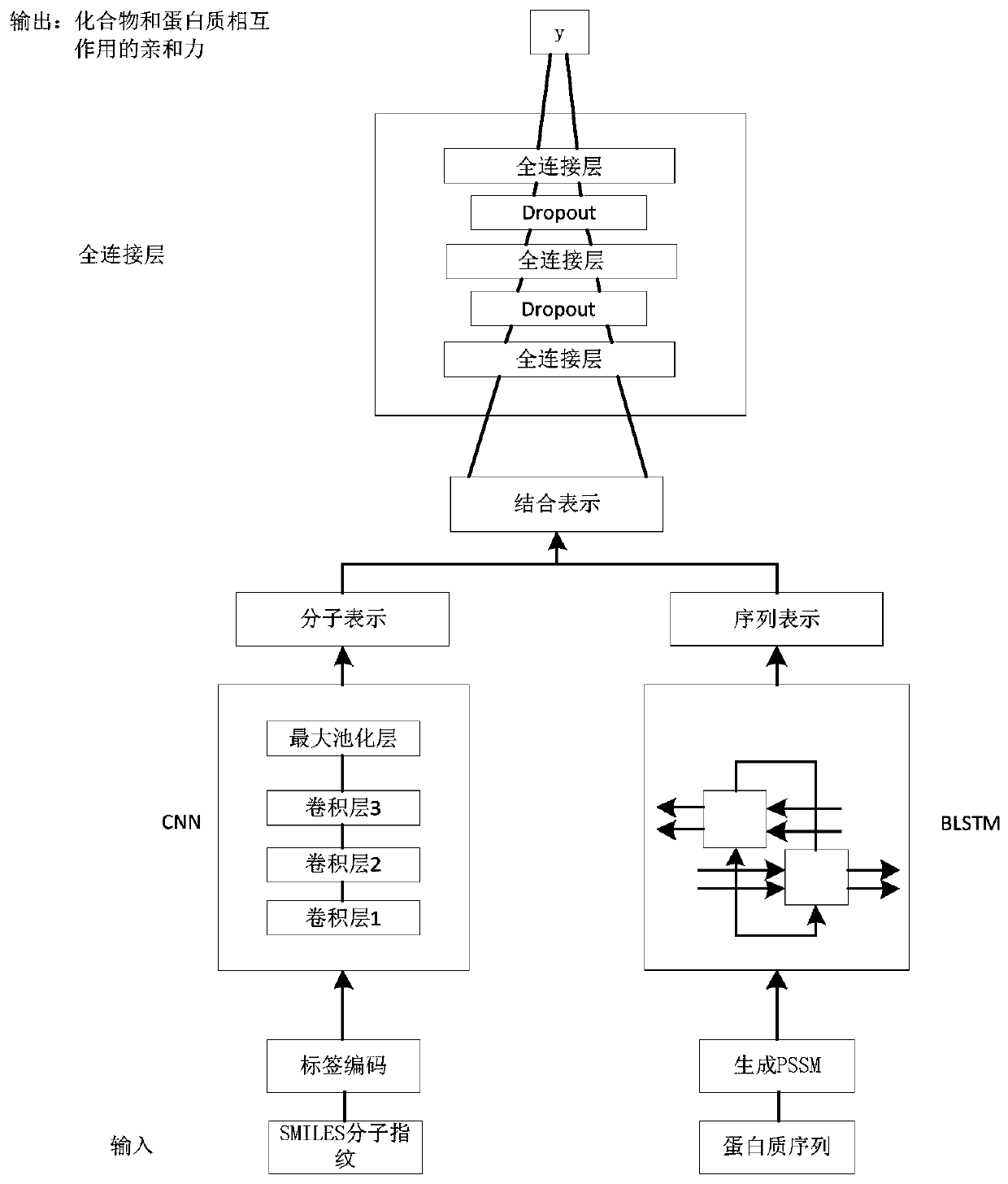
[0044] Please refer to figure 1 with figure 2 , the present invention provides a drug target affinity prediction method based on deep learning, comprising the following steps:
[0045] S1, data preparation, obtain drug compound and target protein data from Davis dataset and KIBA dataset;
[0046] S2. Data processing, encoding the compound, using molecular fingerprints to represent, generating tag codes, performing sequence representation on the protein, and representing the protein using a position-specific scoring matrix;
[0047] S3. Compound feature extraction, constructing a CNN model, inputting label codes into the CNN model, performing feature extraction on the compound, and obtaining the molecular representation of the compound;
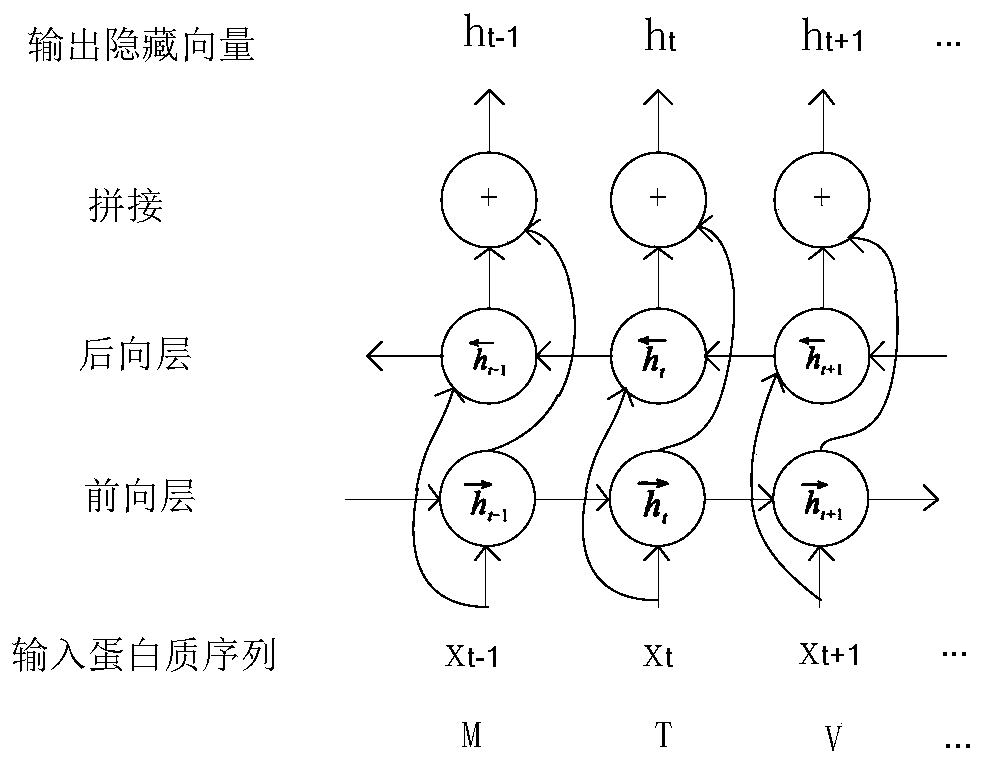
[0048] S4. Protein feature extraction, construct LSTM model, input protein position-specific scoring matrix into LSTM model, perform feature extraction on protein sequence, learn the order relationship between amino acids in protein structu...
Embodiment 2
[0057] For step S2 in Example 1, encoding the compound specifically includes: expressing the chemical structure of each compound as a set of ASCII codes through the SMILES code of the molecule, and each ASCII code represents a substructural feature of the compound.
[0058] In this example, the .mol format file that saves the chemical structure information is downloaded from the TCMSP database, and Openbabel is used to process the .mol format file to calculate the SMILES molecular structure specification of the compound, and then use the "rcdk" package in the R language " get.fingerprint" function can calculate the SMILES code of drug molecules.
[0059] Example of SMILES molecular fingerprint representation of a compound: SMILES molecular fingerprint carbon dioxide 'O=C=O'. Compounds are represented by letters and symbols. For better operation and processing of the algorithm, letters and symbols are converted into numerical forms, integers are used to represent letters an...
Embodiment 3
[0061] For step S2 in Example 1, since there are more than 30 amino acids obtained from natural protein hydrolysis and 20 basic amino acids, each protein is generally composed of these 20 common amino acids, so the position-specific scoring matrix PSSM can represent Be n×20 matrix M={M i→j , i=1...n, j=1...20}, matrix element M i→j Indicates the possibility of the amino acid at the i-th position of an amino acid changing into amino acid j during the evolution process. The larger the number, the higher the possibility of being replaced during the evolution process. n indicates the total number of residues in a given protein sequence .
[0062] In this embodiment, the acquisition of PSSM needs to use PSI-BLAST software to perform amino acid multiple sequence comparison in the nr database (non-redundant protein database) to find homologous sequences, and the formal definition is as follows:
[0063]
[0064] The numbers in each row in the PSSM add up to 1.
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More