

Parallel polymorphism scoring by amplification and error correction
a technology of error correction and parallel polymorphisms, applied in the field of detecting polymorphisms, can solve the problems of error-correcting polymerases, polymerases, polymerases that are ill-suited to amplification of sequences directly from genomic dna, and are difficult or expensive to develop, and achieve the effect of improving the processivity of polymeras
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Benefits of technology
Problems solved by technology
Method used
Image

Examples
example 1
Modified Error-Correcting Enzymes are Superior to Unmodified Error-Correcting Enzymes in Amplifying DNA from Low Copy Number Templates.
[0102] The efficiency with which modified and unmodified error-correcting polymerases can amplify products from small numbers of input template copies was tested using “real-time” PCR. PCR was performed in the presence of the double-stranded-DNA-specific fluorescent dye SYBR Green I (Molecular Probes, Eugene Oreg.) in a DNA Engine Opticon continuous fluorescence detection thermal cycling system (MJ Research, Waltham Mass.). A 57 bp portion of the human cytochrome P450 gene CYP2D6 (GenBank Accession # M33388, nucleotides 3265-3322) was amplified using primers F1 (forward) and R1 (reverse) from a template containing a perfect match to both primers. The number of thermal cycles required for the fluorescence to reach a threshold value (threshold cycle, or Ct) was recorded. The Ct value represents the number of cycles required to generate a detectable a...
example 2
The Error-Correcting Enzymes Pfu and PfS Efficiently Correct Mismatched Labeled Bases during PCR Amplification.
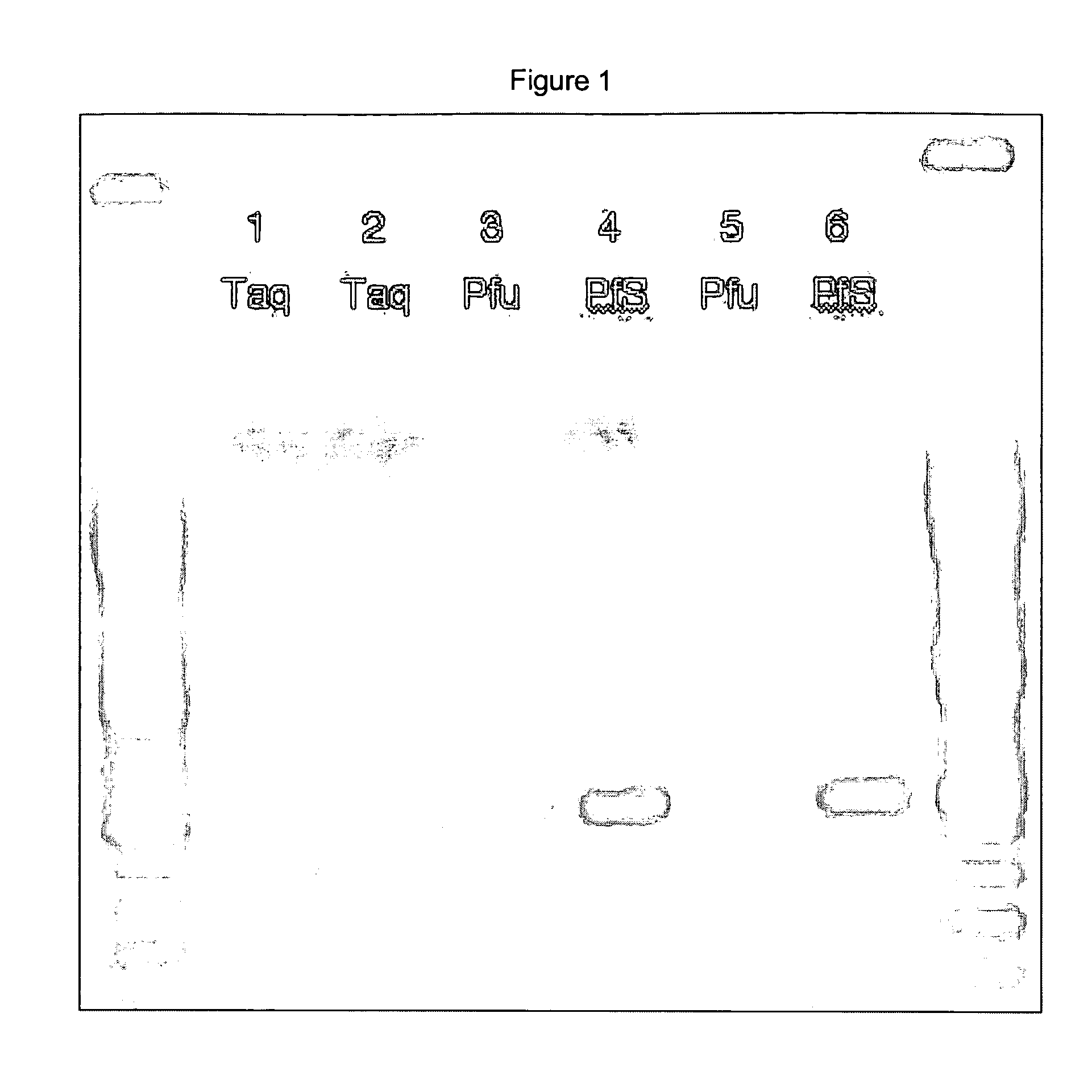
[0118] PCR was performed using a 3′ base-labeled primer in conditions where it had either a perfect match with the template, or a 3′ single-base mismatch with the template. Primer F2 was the base-labeled primer, with the same sequence as primer F1 except the 3′ G (query position) is replaced with a C with a carboxyfluorescein (FAM) dye attached at the 5 position through a linker. 106 copies of plasmid clones with either a G or a C in the polymorphic position were used as templates. The reverse primer, R2, was designed to produce a 475-base amplicon. Enzymes used were Taq Gold (ABI, Foster City Calif.), PfS, and Pfu. Six PCRs were performed, with all combinations of the three enzymes and two templates.
[0119] Conditions were similar to those from example 1. Taq Gold reactions used the commercially-supplied 2× master mix. 2× master mixes were also prepared for Pfu and PfS, ...
example 3
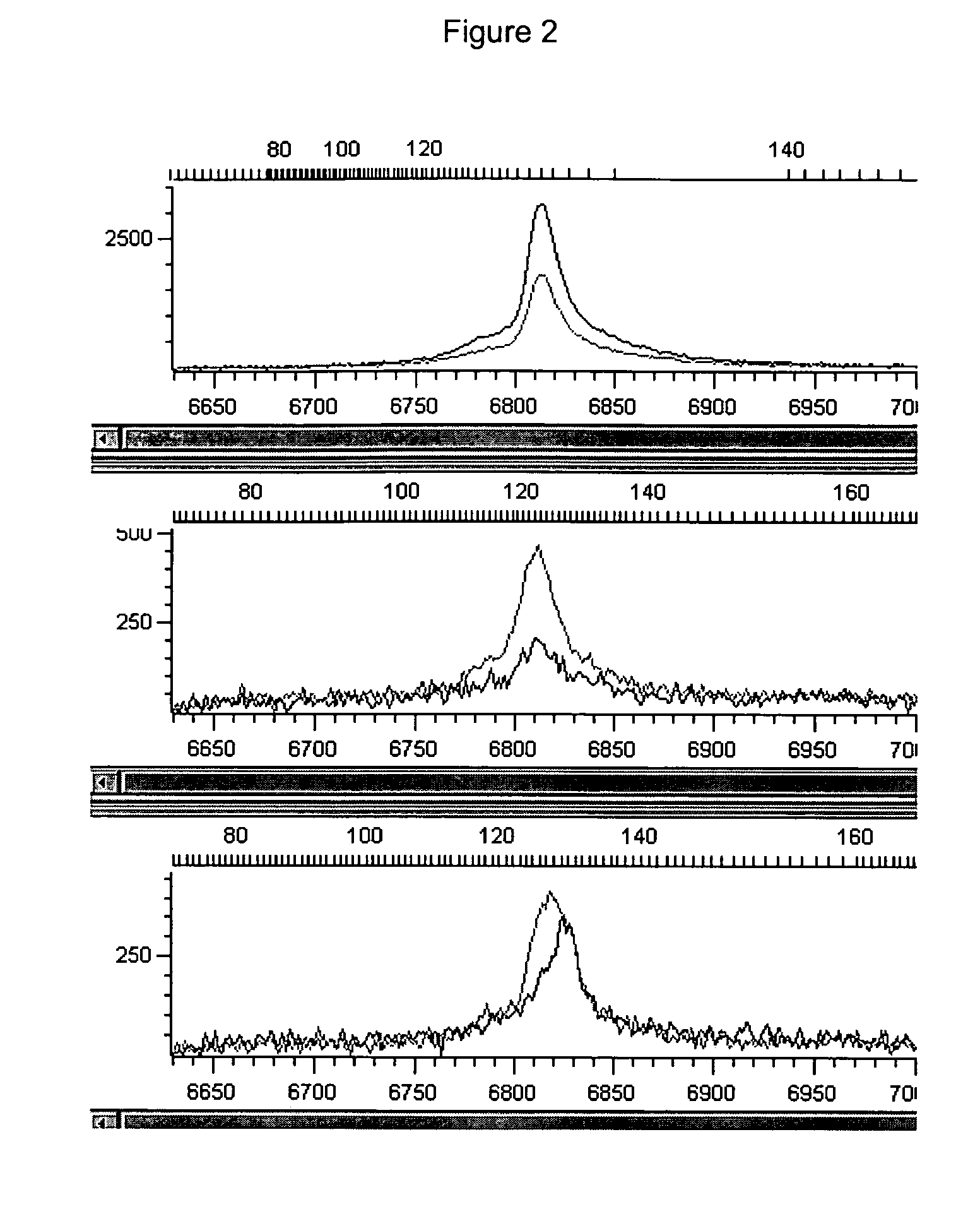
[0132] PCR was performed using a 3′ base-labeled primer in conditions where it had either a perfect match with the template, or a 3′ single-base mismatch with the template. Two labeled primers were used: F2 and F3 were base-labeled, with the same sequence as primer F1 except the 3′ G (query position) is replaced with a C with a carboxyfluorescein (FAM) dye attached at the 5 position through a linker o or a T labeled with Bodipy R6G (BR6G) (Molecular Probes, Eugene Oreg.) linked to the T methyl group via a 6-carbon linker. 106 copies of plasmid clones with either a G or a C in the polymorphic position were used as templates. The reverse primer, R2, was designed to produce a 475-base amplicon. Enzymes used were Taq Gold (ABI, Foster City Calif.), PfS, and Pfu. Twenty seven PCRs were performed, with all combinations of the three enzymes, two primers singly and in combination, and two templates singly and in combination.
[0133] Primer F3 was poorly labeled (only about 7% of molecules we...
PUM
| Property | Measurement | Unit |
|---|---|---|
| temperature | aaaaa | aaaaa |
| temperature | aaaaa | aaaaa |
| temperature | aaaaa | aaaaa |
Abstract
Description
Claims
Application Information
 Login to View More
Login to View More