Heterogenous skin transfected by recombined genes
A skin and wound technology, applied in the field of isolated pig skin, can solve problems such as difficulty, no application, limited effect, etc., achieve good growth conditions, reduce wound scar formation, and promote healing
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image



Examples
Embodiment 1
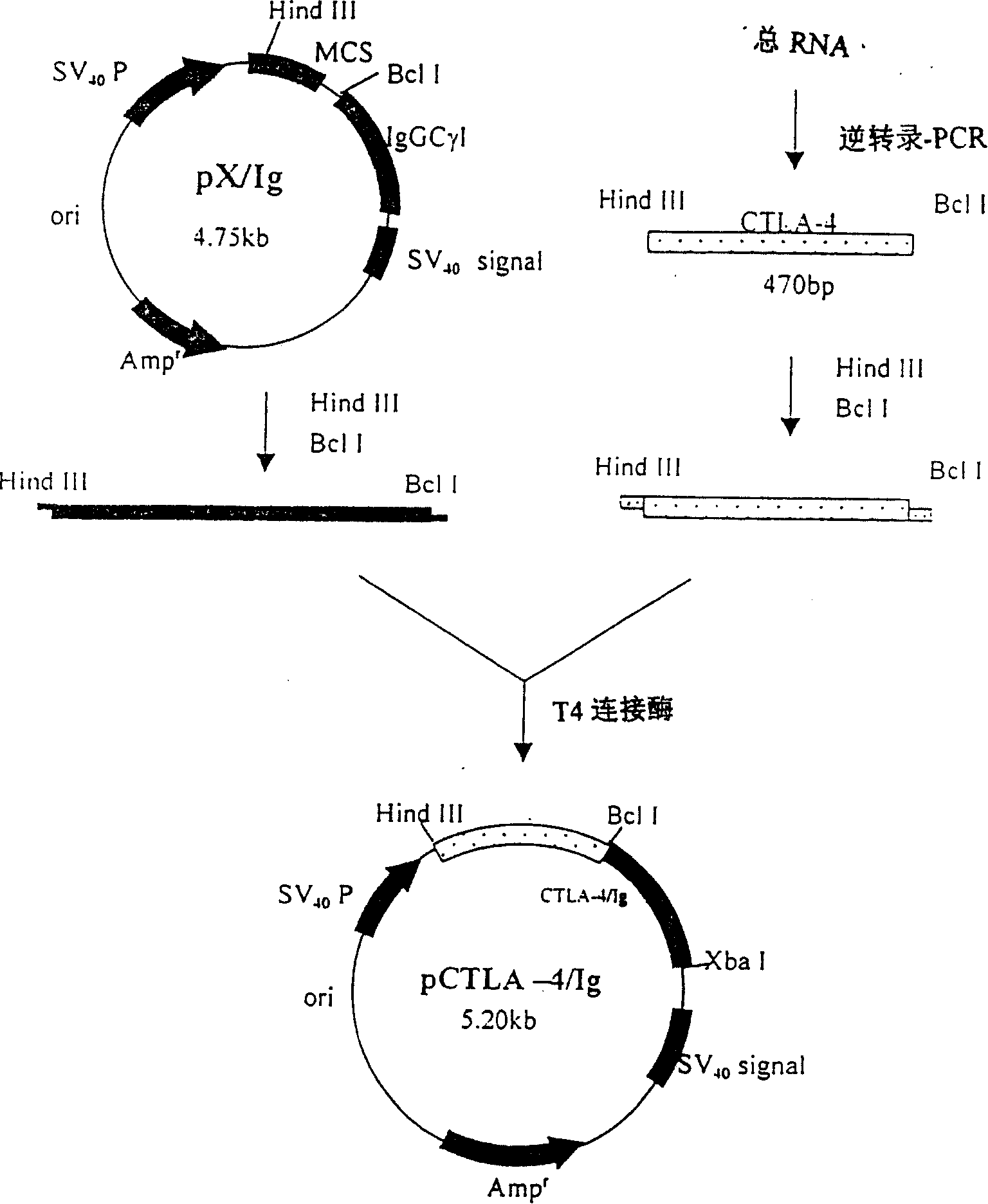
[0022] Construction of Ig fusion protein expression vector ( figure 1 )
[0023] 1. Isolation and activation of peripheral blood mononuclear cells (PBMC)
[0024] Routine isolation of peripheral blood mononuclear cells from healthy people, RPMI1640 adjusted cell density to 1×10 6 Cells / ml, add PMA (15nM) and anti-CD3 monoclonal antibody (10 μg / ml), 37°C 5% CO 2 After culturing for 6 hours, the lymphocytes were activated and CTLA-4mRNA was highly expressed.
[0025] 2. Extraction and identification of total cellular RNA
[0026] 1) Extraction: Collect activated PBMCs by centrifugation, wash cells with PBS (DEPC) × 2, and use 1 × 10 7 Add 1 ml of Tripure to PBMC, mix well, RT for 5 minutes, add 0.2 ml of chloroform, shake vigorously for 15 seconds, RT for 5 minutes, centrifuge at 10,000 rpm for 15 minutes, transfer the upper colorless aqueous phase to another centrifuge tube, add 0.5 ml of isopropanol, mix well, -20°C for 10 minutes, centrifuge at 10,000 rpm for 15 minutes...
Embodiment 2
[0053] The construction of embodiment 2CTLA-4Ig retrovirus expression vector
[0054] pLXSN (Clontech, USA) and pUC19-CTLA-4Ig were digested with EcoRI and HindIII, respectively, and then filled with dATP, dNTP and DNA E. coli polymerase Klenow fragments, and then digested with BamHI, and the electrophoresis The large fragment of pLXSN was recovered and the small fragment of pUC19-CTLA-4Ig was ligated to obtain the expression pLXCTLA-4Ig of CTLA-4Ig.
Embodiment 3
[0055] Example 3 Construction of CTLA4Ig Adenovirus Expression Vector
[0056] 1. Blunt the ends of the cDNA fragments.
[0057] Add 1 μl of 10× buffer solution to the ethanol-precipitated cDNA (0.5 pmol) centrifuge tube, add sterile double distilled water to 9 μl, 70°C for 5 minutes, add 1 μl T4 DNA polymerase, mix gently, do not shake vigorously, and incubate at 37°C for 5 minutes Minutes, adjust the DNA concentration to 1 μg and 50 μl with DNA Dilition buffer, and mix well. Transfer to an ice bath to inactivate the polymerase, which can be used directly in the ligation reaction (if it cannot be used immediately, it should be extracted and precipitated with alcohol immediately, then dissolved in DNA Dilition buffer and stored at -20°C).
[0058] 2. The blunt-ended cDNA was inserted into pAxCAwt (TakaRa, Japan) Koshi plasmid.
[0059] Linearize pAxCAwt: pAxCAwt 5 μl, h buffer (high salt buffer) 5 μl, Swa I 2 μl, ddH 2 O 38μl, mix well and incubate at 25°C for 2 hours; add ...
PUM
| Property | Measurement | Unit |
|---|---|---|
| Titer | aaaaa | aaaaa |
Abstract
Description
Claims
Application Information
 Login to View More
Login to View More - R&D
- Intellectual Property
- Life Sciences
- Materials
- Tech Scout
- Unparalleled Data Quality
- Higher Quality Content
- 60% Fewer Hallucinations
Browse by: Latest US Patents, China's latest patents, Technical Efficacy Thesaurus, Application Domain, Technology Topic, Popular Technical Reports.
© 2025 PatSnap. All rights reserved.Legal|Privacy policy|Modern Slavery Act Transparency Statement|Sitemap|About US| Contact US: help@patsnap.com