

Synthetic method for pharmaceutical intermediate
A synthesis method and intermediate technology, applied in the field of synthesis of pharmaceutical intermediates, can solve problems such as low product conversion rate, harsh reaction conditions, and low production safety, and achieve simple production process, simple purification treatment, and low preparation cost Effect
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

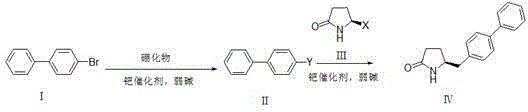
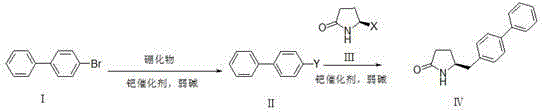
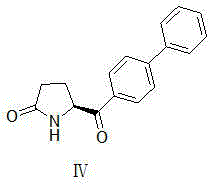
Examples
Embodiment 1
[0038] Dissolve compound Ⅰ (6.99g, 30mmol) in anhydrous DMSO (90ml), add bis-valeryl diboron (15.24g, 60mmol), potassium acetate (12g, 120mmol) and PdCl in sequence 2 (DPPF)-DCM (1.2 g, 1.5 mmol). After the reaction solution was degassed, it was stirred at 80°C overnight (12-20 hours). TLC spots the plate, and the raw material is completely reacted. The reaction solution was filtered through celite, and the filter residue was washed with ethyl acetate (500ml). The filtrate was washed with 10% ammonium chloride solution and saturated brine respectively, the organic phase was dried with anhydrous sodium sulfate, filtered with suction, and the filtrate was concentrated under reduced pressure to obtain 7.9 g of white solid with a yield of 94%.
[0039] Compound III (3.28g, 20mmol, X=Br) was dissolved in a mixture of DMSO (50ml) and water (10ml), and compound II (7.87g, 28.1mmol) and potassium carbonate (11g, 80mmol) were added in sequence. After the reaction solution was degass...
Embodiment 2
[0041] Under nitrogen protection, compound Ⅰ (11.7g, 50.2mmol) was dissolved in anhydrous THF (100ml), cooled to -78°C, and 2.5M n-BuLi solution (24ml, 60.2mmol) was added dropwise, keeping the temperature below - Stir at 70°C for 1h, add trimethyl borate (3.86g, 75.3mmol) dropwise, and stir at -70°C for 2h. TLC spots the plate, and the raw material is completely reacted. Slowly lower to room temperature, add 10% hydrochloric acid solution (40ml) dropwise, and stir for 30min. Stand still, separate the organic layer, extract the aqueous layer with ethyl acetate (100ml*3), combine the organic phases, dry the organic phase with anhydrous sodium sulfate, filter with suction, and concentrate the filtrate under reduced pressure to obtain 8.96 g of solids with a yield of 90.1 %.
[0042] Compound Ⅲ (3.28g, 20mmol, X=Br) was dissolved in a mixture of DMSO (50ml) and water (10ml), and compound Ⅱ (5.56g, 28.1mmol), potassium carbonate (11g, 80mmol), PdCl 2 (DPPF)-DCM (0.8 g, 1 mmol)....
Embodiment 3
[0044] Compound Ⅰ (6.99g, 30mmol) was dissolved in anhydrous DMSO (90ml), and dimethylpropanediol diboronic acid ester (15.97g, 60mmol), potassium acetate (12g, 120mmol) and PdCl were added successively 2(DPPF)-DCM (1.2 g, 1.5 mmol). After the reaction solution was degassed, it was stirred at 80°C overnight (12-20 hours). TLC spots the plate, and the raw material is completely reacted. The reaction solution was not processed and directly entered into the next reaction.
[0045] Compound III (3.28g, 20mmol, X=Br) was dissolved in a mixture of DMSO (50ml) and water (10ml), and the reaction solution obtained in the previous step and potassium carbonate (11g, 80mmol) were added in sequence. After the reaction solution was degassed, the temperature was raised to 80°C and stirred overnight (12-20 hours). TLC spots the plate, and the raw material is completely reacted.
[0046] The reaction solution was filtered through celite, and the filter residue was washed with ethyl acetate...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More