

Method for preparing edoxaban chiral amine intermediate
A technology of intermediates and compounds, applied in the field of medicinal chemistry, can solve problems such as unfavorable production safety, achieve cost reduction, reduce the risk of residual nitrosamine carcinogenic compounds, and reduce the possible effects of nitrosamine compounds
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
Embodiment 1
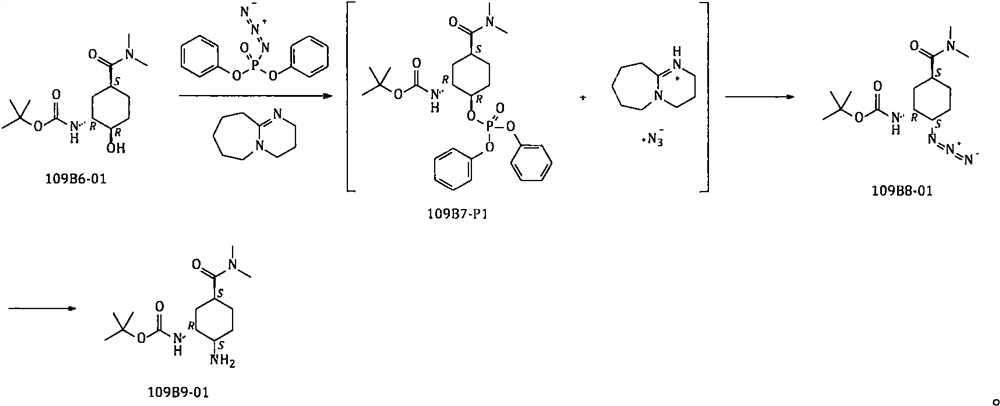
[0043] Example 1 Synthesis of N-[(1R, 2R, 5S)-5-[(dimethylamino)carbonyl]-2-hydroxycyclohexyl]carbamate tert-butyl ester
[0044]
[0045]Add 109B4-01 (1200g; 7.091mol) to the reaction flask, add concentrated ammonia water (6000g), heat to 40°C and react for 8-10hrs. Concentrate under reduced pressure until about 2000-2500 g remains in the reaction flask. Add pre-prepared and cooled to room temperature sodium hydroxide aqueous solution (a solution of 600g sodium hydroxide and 5400g water), keep warm at about 40°C, add Boc-anhydride (1920g; 8.797mol) in batches; after adding, keep warm at React at 40-50°C for 2-3 hours. After cooling, dichloromethane (4800g) was added for extraction, and the aqueous phase was extracted with dichloromethane (1200g×2). Combine the organic phases; dry with anhydrous sodium sulfate, filter, and concentrate the filtrate; add 6000 g of toluene to the obtained residue, heat and stir to disperse for 1 to 2 hours, and cool to crystallize. The soli...
Embodiment 2
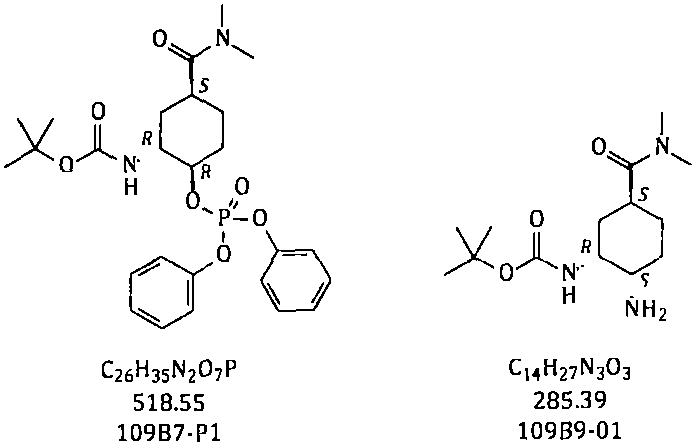
[0046] Example 2 Synthesis of N-[(1R, 2S, 5S)-2-azido-5-[(dimethylamino)carbonyl]cyclohexyl]carbamate tert-butyl ester
[0047]
[0048] Add toluene (3000g) to the reaction flask, then add 109B6-01 (600g; 2.095mol), add DBU (420g; 2.759mol), add diphenylphosphoryl azide (DPPA) (750g; 2.725mol). Heat to 45-50°C for 2-3hrs, then add anhydrous potassium carbonate (500g; 3.618mol), continue to heat up to 100-105°C, and stir for 36-40hrs.
[0049] After the reaction is completed, cool to 40-50°C, add water (3000g) to the reaction system, keep warm at 40-45°C, stir and extract, keep the water phase at 40-45°C, and extract with toluene three times (1200g+600g× 2); combine the organic phases; dry with anhydrous sodium sulfate, filter, collect the filtrate, concentrate toluene under reduced pressure to obtain a residue; add ethyl acetate and n-heptane (1:3, w / w) to the residue , stirred and crystallized at room temperature; filtered, collected the solid, and dried to obtain 109B8-0...
Embodiment 3
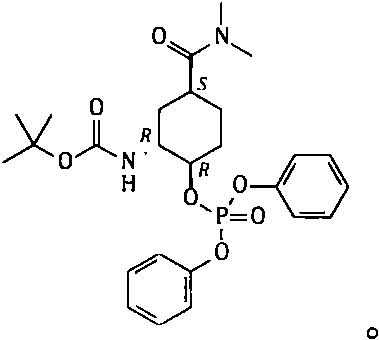
[0050] Example 3 Synthesis of N-[(1R, 2R, 5S)-5-[(dimethylamino)carbonyl]-2-[(diphenoxyphosphoryl)oxy]cyclohexyl]carbamate tert-butyl ester
[0051]
[0052] Add toluene (800g) to the reaction flask, then add 109B6-01 (100g; 349.2mmol), add DBU (75g; 492.6mmol), add diphenylphosphoryl azide (DPPA) (150g; 545.1mmol). Heat to 45-50°C, keep warm for 4 hours, cool to room temperature, stir and crystallize; filter, collect the solid, and dry to obtain 109B7-P1 with a dry weight of about 157g. Yield: 86.7% (theoretical amount: 181.08 g).
[0053] Take part of the solid obtained above, heat beating and refining with toluene to obtain purified 109B7-P1, and its nuclear magnetic spectrum data are as follows:
[0054] 1 H-NMR (500MHz, CDCl 3 ): 1.40ppm(s, 9H); 1.52~2.19ppm(m, 2H+2H+2H); 2.78ppm(m, 1H); 2.92ppm, 3.00ppm(d, 3H+3H); 4.10~4.12ppm( m, 1H); 4.66 ~ 4.67ppm (m, 1H); 5.67pm (br, 1H); 7.16 ~ 7.18ppm (t, 2H); 7.21 ~ 7.25ppm (m, 4H); 4H).
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More