

Third-generation full-length transcriptome sequencing result analysis method suitable for Sequel sequencing
A transcriptome analysis, full-length technology, applied in genomics, proteomics, instruments, etc., can solve the problems of slow running time and high computer resource consumption, and achieve fast running speed, easy analysis, and fine annotation
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
Embodiment Construction
[0057] A three-generation full-length transcriptome analysis method applicable to the Sequel sequencing platform, comprising the following steps:
[0058] Step 1, sequencing data filtering step:
[0059] Use pacbio's official isoseq3 process to process the raw data:
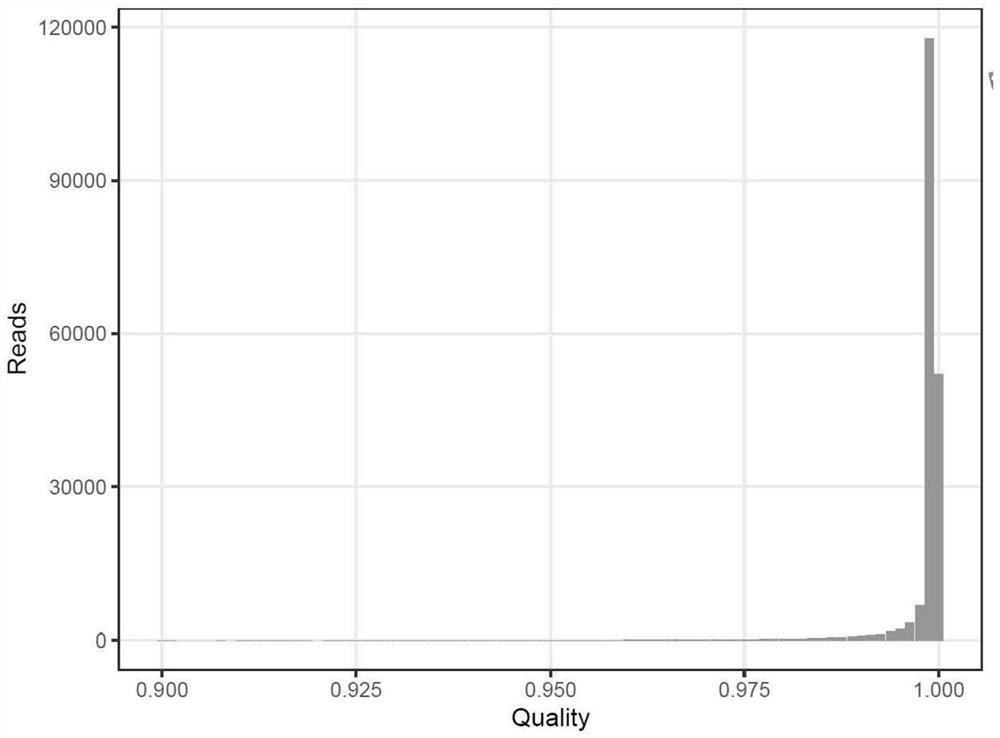
[0060] Use the ccs program to process the off-machine subreads to obtain the consistency sequence CCS of each zero-mode waveguide hole, as shown in figure 1 As shown, the accuracy value distribution of pacbio CCS (Consensus Sequence) is mainly distributed around 0.99, indicating that the sequencing results are of very high quality after processing;
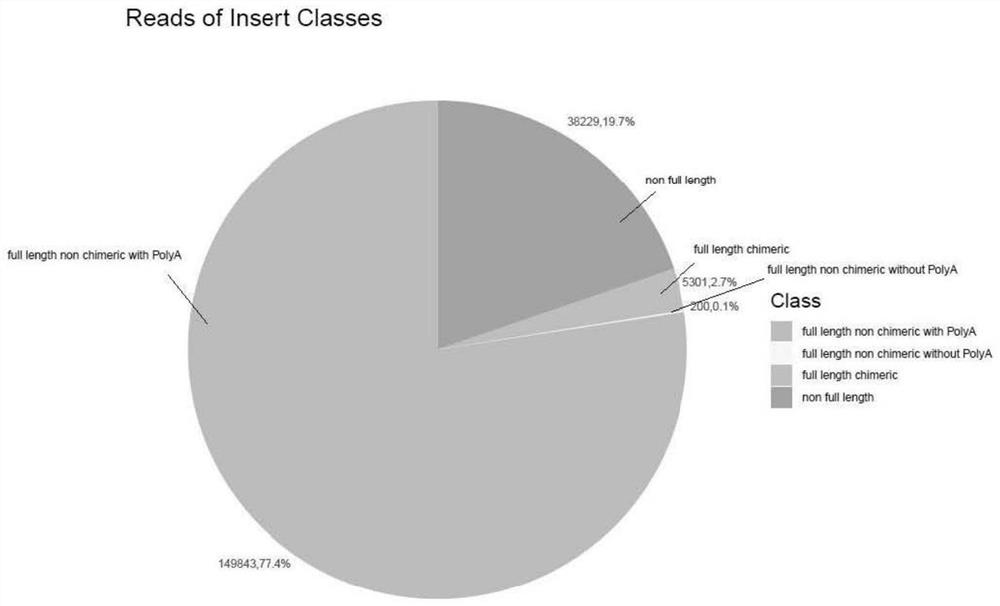
[0061] Use the lima program to identify the joints of the consensus sequence to obtain the full-length sequence FL, as shown in figure 2 As shown in the figure, the sequences of full length non chimeric with PolyA (full length non-chimeric, containing PolyA) account for the vast majority, and the effective sequence comparison in the result is relatively high;
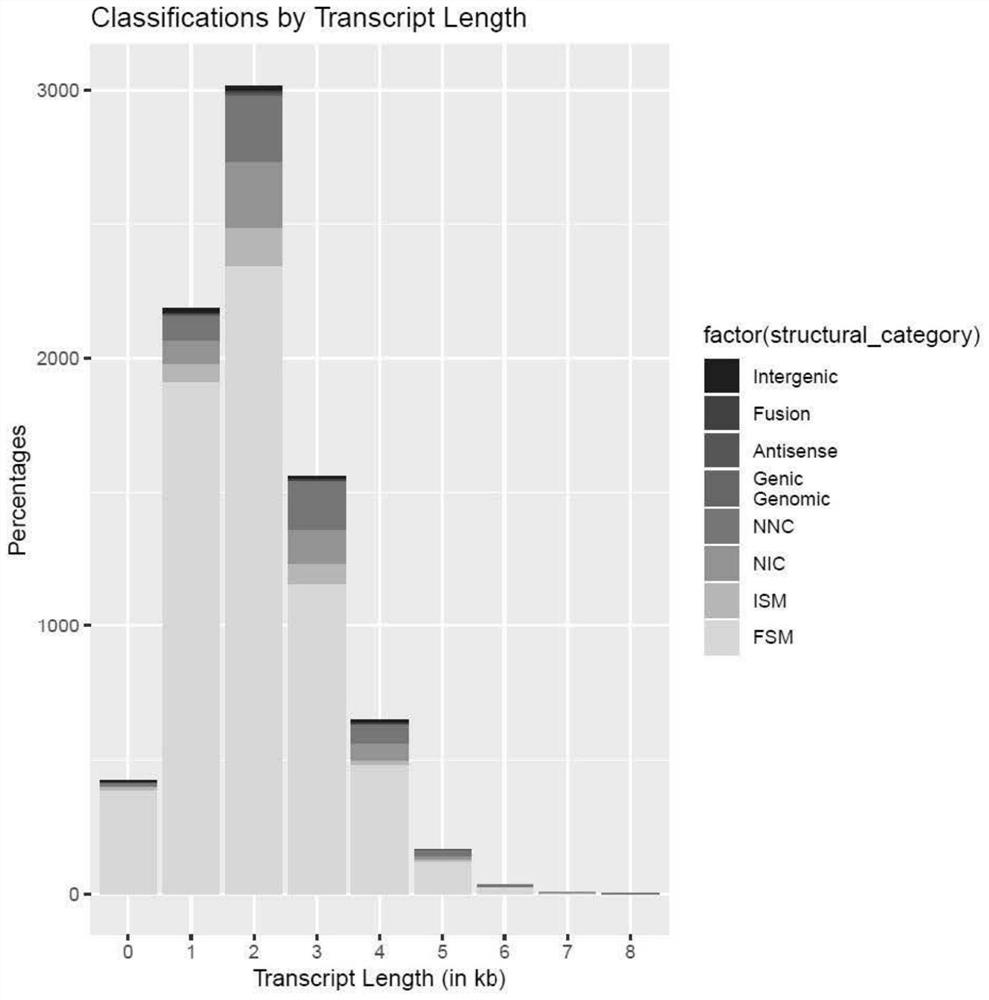
[00...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More