

Pharmaceutical product comprising a muscarinic receptor antagonist and a beta2-adrenoceptor agonist
A technology of receptor agonists and pharmaceutical products, applied in the direction of medical preparations containing active ingredients, drug combinations, pharmaceutical formulations, etc., can solve problems such as unsatisfactory drug efficacy
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
preparation example Construction
[0164] Preparation of muscarinic antagonists
[0165] The muscarinic antagonists of the present invention can be prepared as follows. Alternative salts to those described in this application can be prepared by conventional chemistry using methods analogous to those described.
[0166] General Experimental Details for the Preparation of Muscarinic Antagonists
[0167] Unless specifically stated otherwise, the following general conditions were used in the preparation of muscarinic antagonists.
[0168] All reactions were performed under nitrogen atmosphere unless otherwise specified.
[0169] In the examples, NMR spectra were measured on a Varian Unity Inova spectrometer at a proton frequency of 300 or 400 or 500 MHz, or on a Bruker DRX spectrometer at a proton frequency of 400 or 500 MHz, or on a Bruker Avance spectrometer at a proton frequency of 600 MHz The measurements were made at a proton frequency of 300 MHz or on a Bruker Avance DPX 300 spectrometer. MS spectra wer...
Embodiment 1
[0199] Example 1: (R)-3-(1-phenyl-cycloheptanecarbonyloxy)-1-(pyrazin-2-ylcarbamoylmethyl)-1-nitrogen - Bicyclo[2.2.2]octane bromide
[0200] a) 1-Phenyl-cycloheptanol
[0201]
[0202] To a solution of magnesium (1.2 g) in anhydrous tetrahydrofuran (60 mL) was added crystals of iodine followed by bromobenzene (7.85 g) under nitrogen at such a rate that the reaction maintained a steady reflux. The reaction mixture was stirred for 20 minutes then cycloheptanone (4.48g) was added carefully. After stirring for 10 minutes, saturated aqueous ammonium chloride (10 mL) was added and the reaction mixture was partitioned between water (100 mL) and isohexane (100 mL). The organic layer was dried (MgSO 4 ) and evaporated to give the subtitle compound (7.6 g) as an oil.
[0203] 1 H NMR (299.946MHz, CDCl 3 )δ7.53-7.47(m, 2H), 7.36-7.29(m, 2H), 7.26-7.19(m, 1H), 2.07(ddd, 2H), 1.97-1.50(m, 11H).
[0204] b) 1-methoxy-1-phenyl-cycloheptane
[0205]
[0206] 1-Phenyl-cyclohept...
Embodiment 1
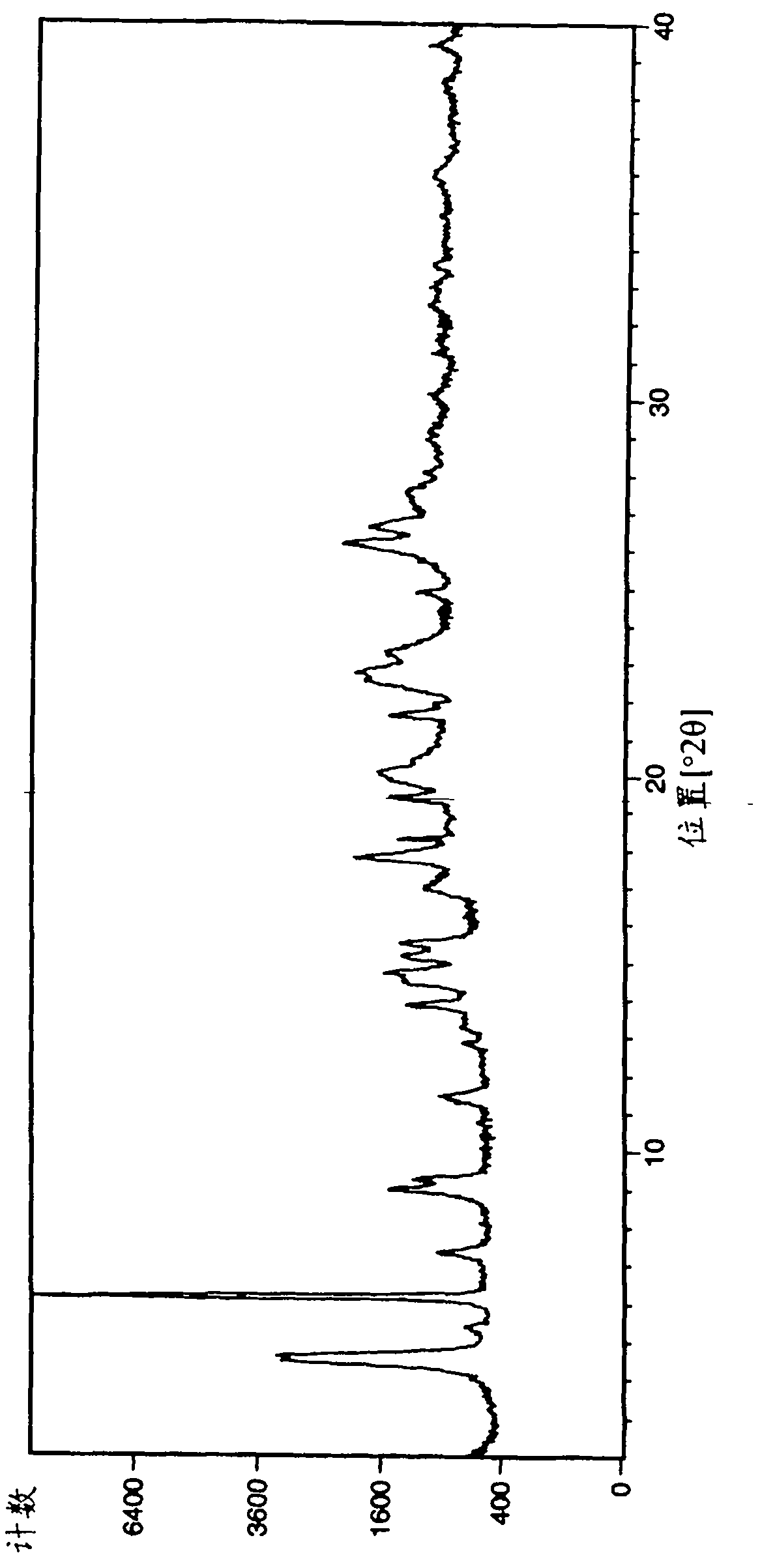
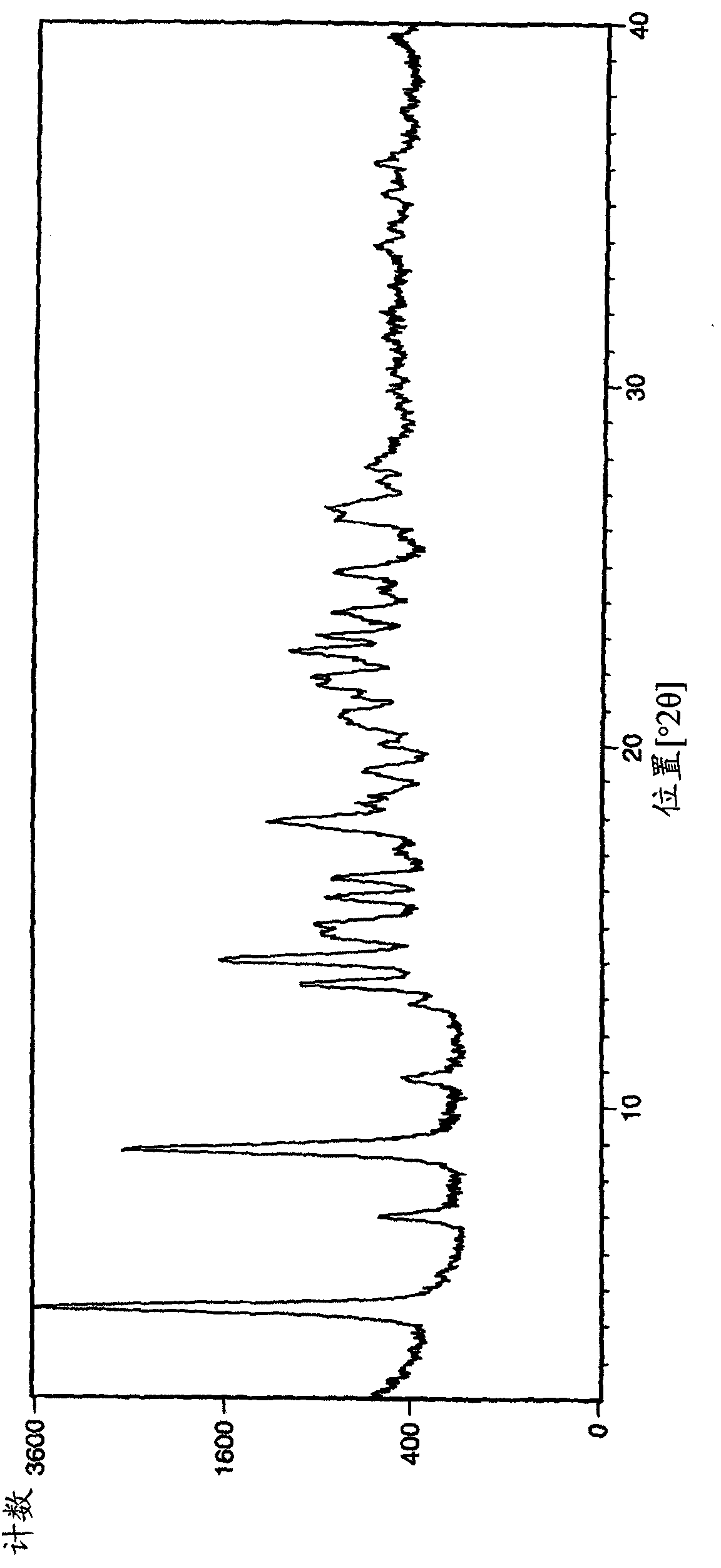
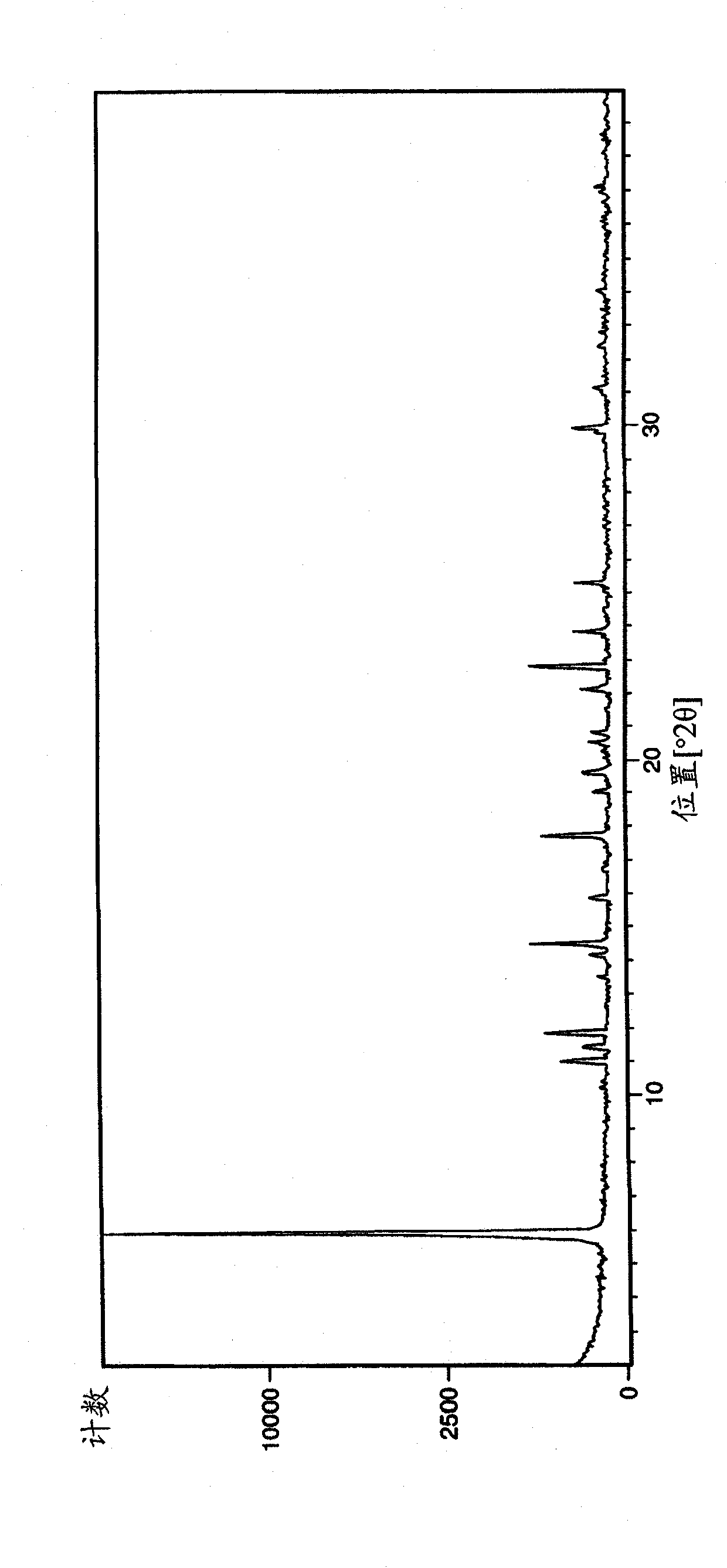
[0230] For Example 1: (R)-3-(1-phenyl-cycloheptanecarbonyloxy)-1-(pyrazin-2-ylcarbamoylmethyl)-1-nitrogen -Analysis of bicyclo[2.2.2]octane bromide crystal form A
[0231] The samples of crystal form A of Example 1 obtained by the above operation were analyzed by XRPD (PANalytical X'Pert or CubiX system), DSC and TGA.
[0232] The melting temperature (melting temperature) of bromide crystal form A of Example 1 determined by DSC was measured to be 202°C (initial) (±2°C). The observed weight loss by TGA prior to melting was 2.7%. GVS was determined to give a 3% weight gain (% w / w) at 80% RH (±0.2%).
[0233] The XRPD spectrum of embodiment 1 bromide crystal form A is in figure 1 displayed in .
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More