

Method for preparing acotiamide hydrochloride
The technology of a compound and an organic base is applied in the field of preparing the gastric motility drug acotiamide hydrochloride, which can solve the problems of high price, troublesome operation, unfavorable industrial production and the like, and achieve the effects of simple operation and low cost.
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

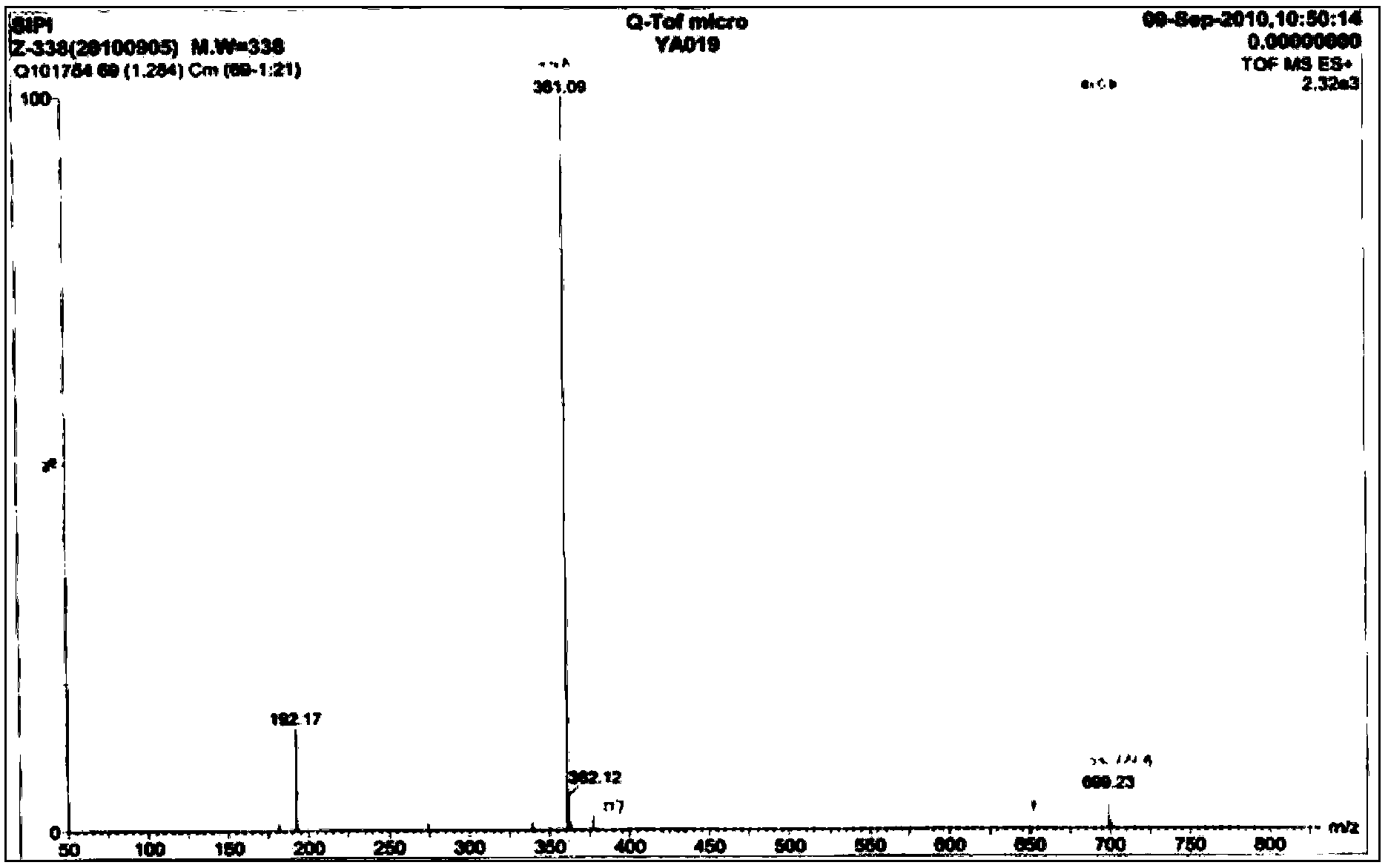
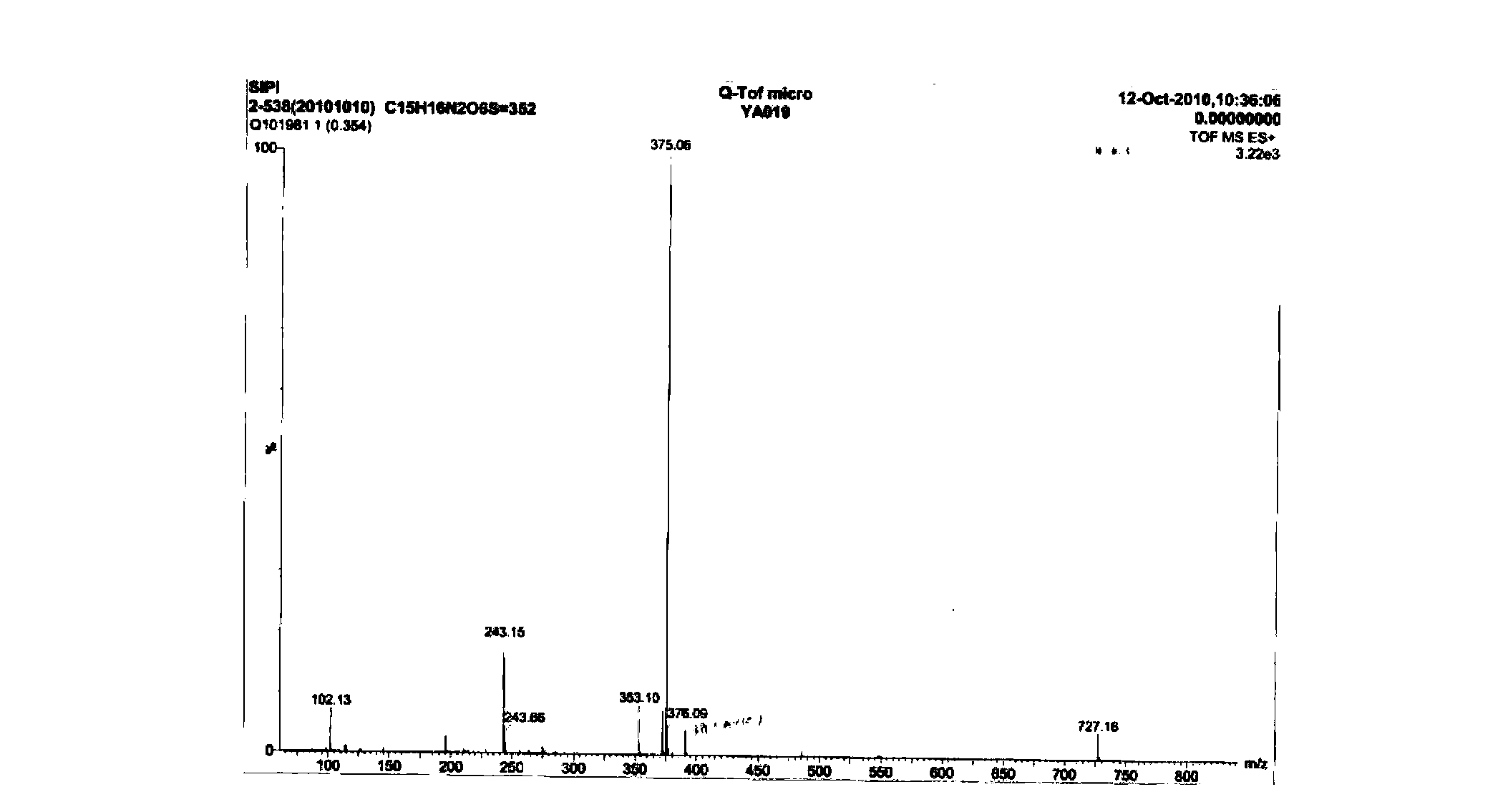
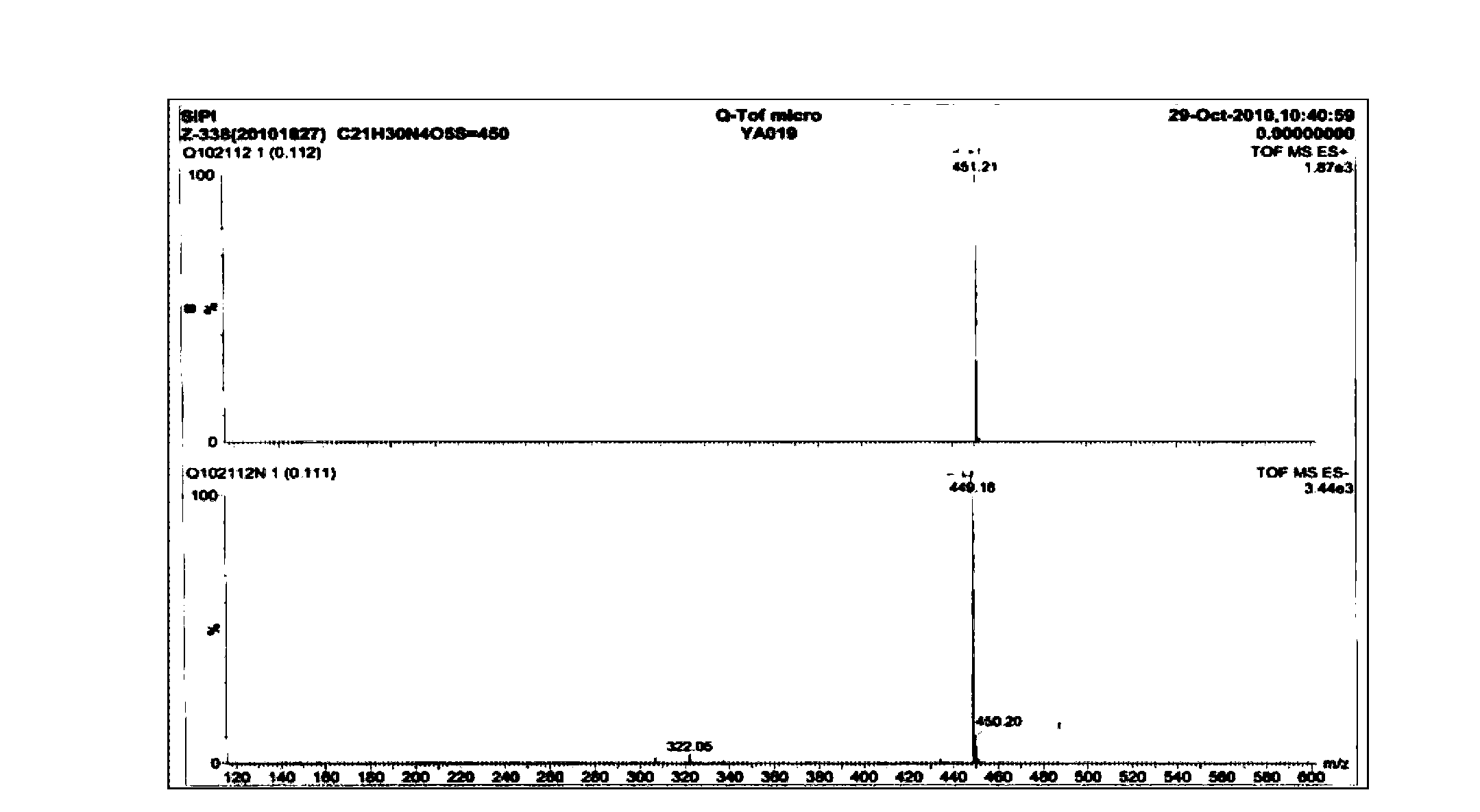
Examples
Embodiment 1
[0029] Synthesis of 2-[(2-hydroxy-4,5-dimethoxybenzoyl)amino]-1,3-thiazole-4-carboxylic acid methyl ester
[0030] Dissolve 19.0 g of triphosgene in 90 ml of CH 2 Cl 2 Placed in a four-necked bottle, N 2 Under air flow, 2-hydroxy-4,5-dimethoxybenzoic acid (22.2 g) was dissolved in 150 ml CH 2 Cl 2 and 45ml of pyridine, drop it into a four-neck flask under an ice-salt bath and control the temperature at 0-5°C. The dropwise addition was completed within 45 minutes, and kept stirring at low temperature for 10 minutes. Then rise to room temperature (20°C) and stir for 50 minutes to stop the reaction. Suction filtration under normal pressure, and the filtrate was rotary evaporated at room temperature to constant weight, 35g of methyl 2-aminothiazole-4-carboxylate and 240ml of 1,2-dichloroethane were added, heated to reflux, and reacted for 6h. After stopping, cool down and filter with suction. The resulting solid was washed with 40 ml of methanol under reflux, and filtered wh...
Embodiment 2
[0032] Synthesis of 2-[(2-hydroxy-4,5-dimethoxybenzoyl)amino]-1,3-thiazole-4-carboxylic acid methyl ester
[0033] Dissolve 3.0 g of triphosgene in 15 ml of CH 2 Cl 2 Placed in a four-necked bottle, N 2 Under air flow, 2-hydroxy-4,5-dimethoxybenzoic acid (3.0 g) was dissolved in 30 ml CH 2 Cl 2 and 6ml of pyridine, drop it into a four-neck flask under an ice-salt bath and control the temperature at 0-5°C. After 20 minutes of dropwise addition, keep stirring at low temperature for 1 hour. Then it was raised to room temperature (20°C) and stirred overnight, and the reaction was stopped after 24 hours. Rotary steam at room temperature to constant weight, add 3.5g methyl 2-aminothiazole-4-carboxylate and 30ml 1,2-dichloroethane, heat to reflux, react for 6h. After stopping, evaporate the solvent to dryness, add 30ml of methanol to reflux and filter to obtain 4.1g of white solid, evaporate the mother liquor to dryness, add 20ml of methanol to wash and filter to obtain 0.85g o...
Embodiment 3
[0035] Synthesis of 2-[(2-hydroxy-4,5-dimethoxybenzoyl)amino]-1,3-thiazole-4-carboxylic acid methyl ester
[0036] Dissolve 3.0 g of diphosgene in 15 ml of CH 2 Cl 2 Placed in a four-necked bottle, N 2 Under air flow, 2-hydroxy-4,5-dimethoxybenzoic acid (3.0 g) was dissolved in 30 ml CH 2 Cl 2 and 6ml of pyridine, drop it into a four-neck flask under an ice-salt bath and control the temperature at 0-5°C. After 20 minutes of dropwise addition, keep stirring at low temperature for 1 hour. Then it was raised to room temperature (20°C) and stirred overnight, and the reaction was stopped after 24 hours. Rotary steam at room temperature to constant weight, add 3.5g methyl 2-aminothiazole-4-carboxylate and 30ml 1,2-dichloroethane, heat to reflux, react for 6h. After stopping, the solvent was evaporated to dryness, 30ml of methanol was added to reflux and suction filtered to obtain 4.57g of white solid, with a yield of 89.6%.
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More