

MHC completion database, and establishment method and application thereof
A construction method and database technology, applied in the field of MHC complete database, can solve problems such as false positive bais in comparison results, highly polymorphic linkage disequilibrium, easy to miss pathogenic sites, etc., to reduce CPU and memory usage , Improve the accuracy of data and the effect of accurate haplotype information
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
Embodiment 1
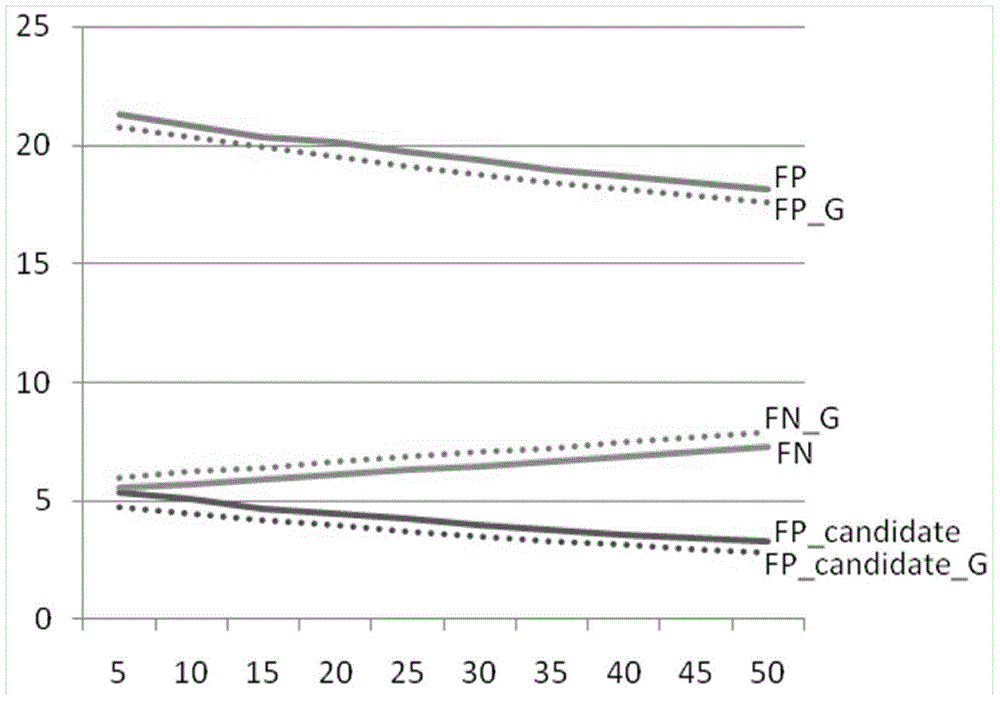
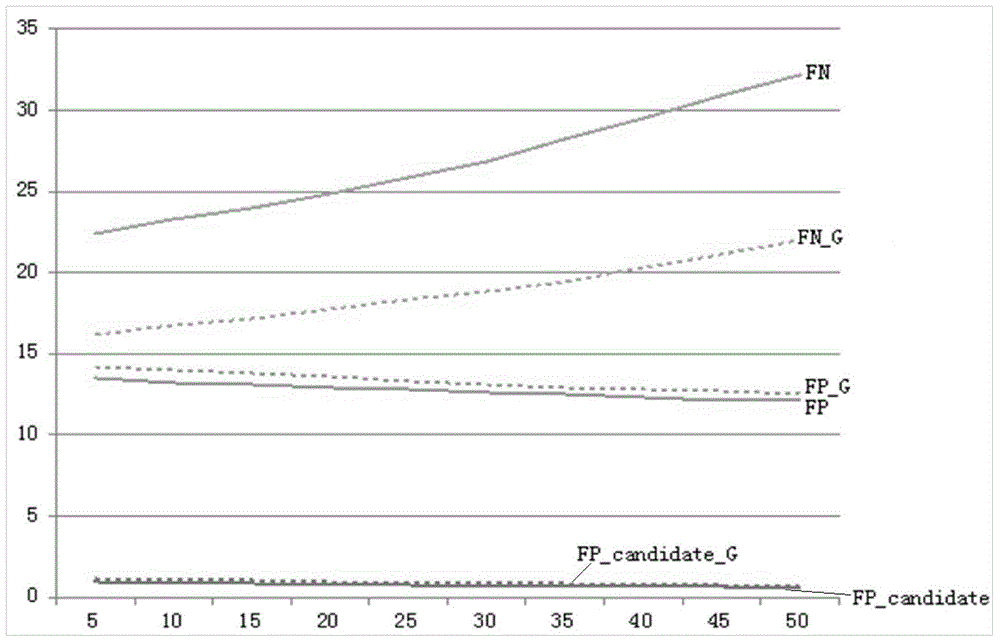
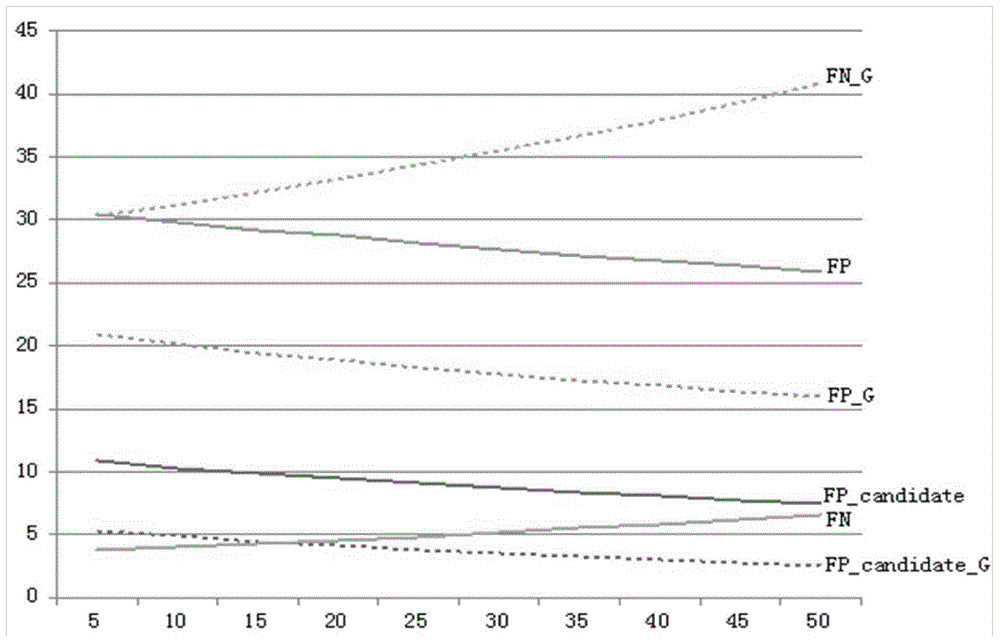
[0044] Embodiment 1 Raw data preparation and variation detection software comparison
[0045] In this example, 8,906 Chinese genomic DNA samples were selected from the BGI project, and the MHCcapture chip was used to capture the sequence of the human MHC region, and the sequence of the captured MHC region was sequenced.
[0046] The original data obtained during the sequencing process in this example is stored in the fastq format file, referred to as the fq format, which stores the read length sequence, that is, the reads are also called reads, and the sequencing quality of the reads and other information. After obtaining the original fq format data, perform basic processing such as removing joints and removing low-quality reads. In this example, the basic processing method adopted by the second-generation sequencing data is used. After the basic processing, a clean sequence, that is, cleanreads, is obtained. The cleanreads are the sequencing results. It should be noted that ...
Embodiment 2
[0053] Example 2 Data filtering and genotype data set
[0054] In this example, the genotype data set in the MHC completion database is constructed according to the variation detection results in Example 1. The genotype data set includes: accurate genotype sites of all samples, including single nucleotide polymorphism sites of the compared populations Point SNPs and insertion deletion polymorphic sites INDELs information.
[0055] In Example 1, we have obtained the genotype result of each sample, that is, the result of variation detection. We use the merge program to extract the genotype result of each sample and cut it into a file to obtain the original genotype of all samples data set.
[0056] And filter the original genotype dataset according to the following three conditions:
[0057] a. Sites with sequencing depth ≥ 6 in the population;
[0058] b. Sites where the missing rate of data in the population is <0.05;
[0059] c. A site where the allelic base type occurs m...
Embodiment 3
[0067] Embodiment 3 MHC completion database
[0068] 1. genotype data set
[0069] In Example 2, we obtained the genotype data set, but the storage format is genotype format. We use GTOOLS software to convert the genotype file into ped and map formats that PLINK can recognize. The parameters are as follows: gtool-G--gsample.gen--ssample.sampleinfo--pedgenotype.ped--mapgenotype.map--snp
[0070] 2. The type data set of HLA typing and the amino acid change information data set corresponding to the typing
[0071] Based on the high-depth reads sequence of each sample, we use the SOAPHLA typing software developed by BGI to perform HLA typing on each sample, obtain the type result of each sample, and store it in ped and map formats, namely HLA A typed dataset of types. For the type results, we find the SNP corresponding to each type based on the IMGT database, and compare it with the SNP at the same position in the human gene standard sequence hg18. If the two are different, we ...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More