Bacteria ncRNA prediction method based on Illumina transcriptome sequencing data and PeakCalling method
A technology of transcriptome sequencing and prediction methods, applied in the field of bioinformatics
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

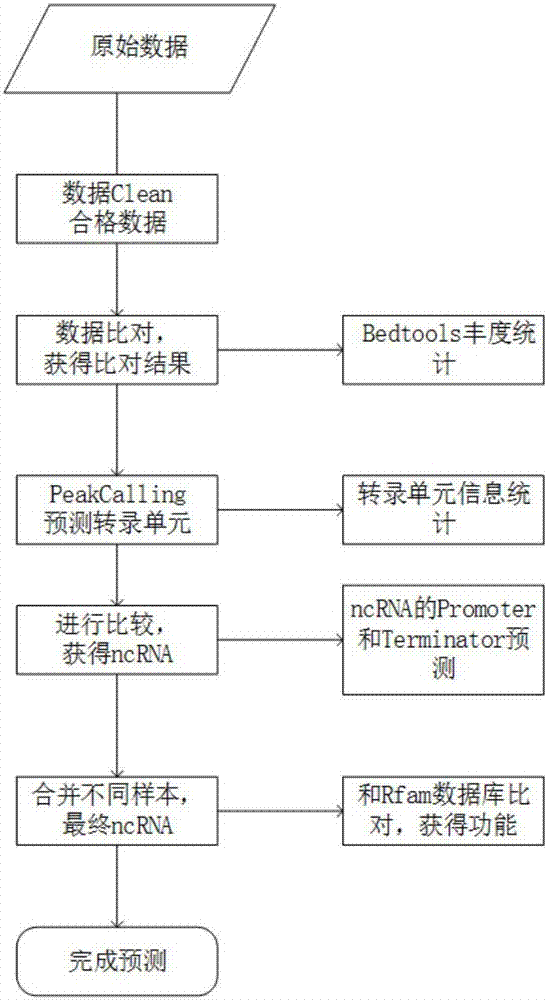
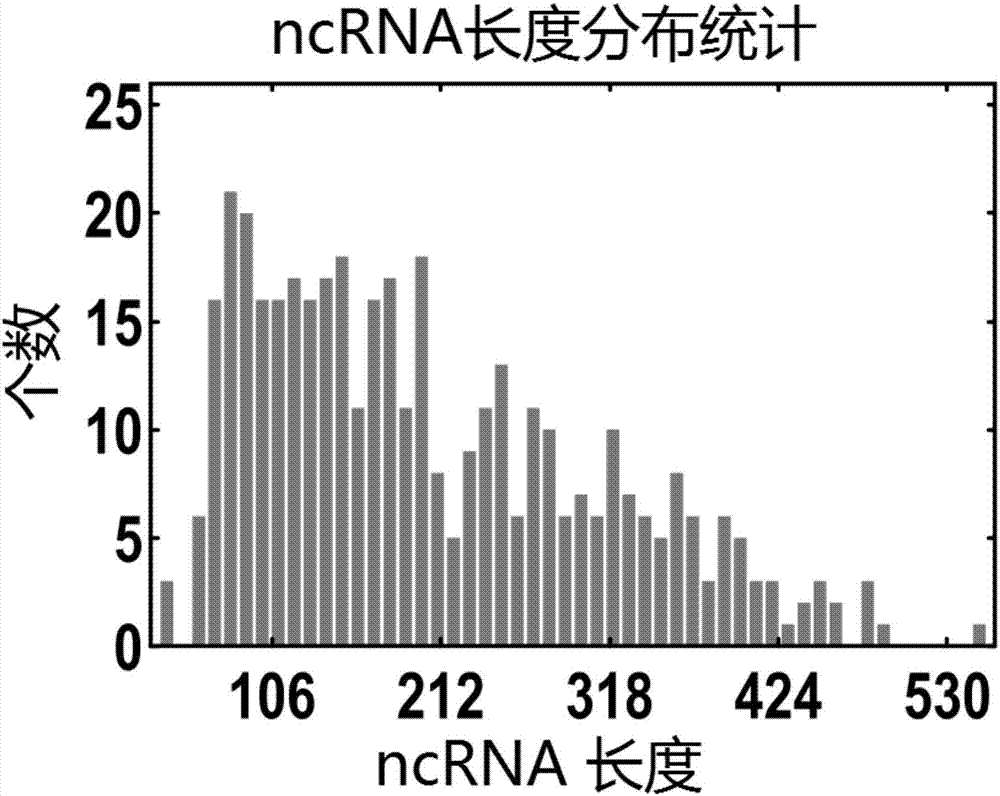
Examples
Embodiment 1
[0043] [Example 1] Obtaining the data to be analyzed of each transcriptome
[0044] We obtained the transcriptome data of a Yersinia pestis, the specific species information is Yersinia Pestis, and there are two transcriptome data: wild strain and knockout strain. For its RNA, first delete the rRNA inside, and then build the library, and then obtain the original sequencing data of the transcriptome of the two groups of rRNA deletion of a certain Yersinia pestis through the Illumina sequencing platform;
[0045] After obtaining the transcriptome sequencing data, filter the unqualified data in each group of raw sequencing data, the unqualified data includes: low-quality reads, wherein the low-quality reads include, more than 30% of the bases in the sequence are of low quality reads of less than 20; reads with a sequence length of less than 16 after trimming off the sequencing adapter; trimming of reads with a sequence length of less than 16 after low-quality base sequences, wher...
Embodiment 2
[0047] [Example 2] Peakcalling method to predict ncRNA of Yersinia pestis
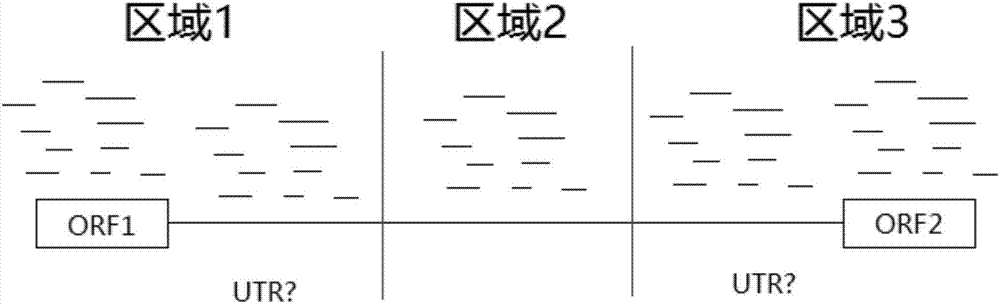
[0048] Use the PeakCalling method to obtain the Peak schematic diagram, see figure 2 .
[0049] 1. Aligning the data to be analyzed of the two transcriptomes to the reference genome of Yersinia pestis;
[0050] Use bowtie or bowtie2 software for comparison. When using bowtie software for comparison, the specific parameters are set as follows: use -v parameter, allow 2 mismatches, and output 2 best matching results; when using bowtie2 for comparison, -N parameter Select 1, use --end-to-end comparison mode, set the number of threads when the program is running to 1~16; set the output file type to sam format.
[0051] 2. After obtaining the comparison results, use the genomecov method of bedtools to count the comparison depth of each position in the reference genome of each sample, perform quantitative analysis on the comparison depth of the whole genome, and select the -d parameter.
[0052] 3. Accor...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More