

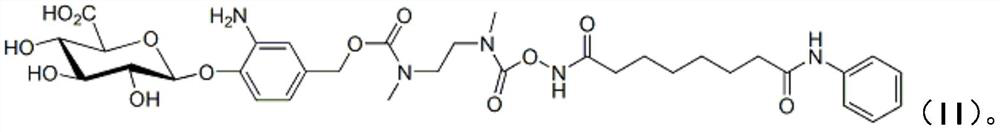
A kind of vorinostat derivative based on palladium carbon and its preparation method and application
A technology of vorinostat and derivatives, which is applied in the field of palladium-carbon-based vorinostat derivatives and their preparation, can solve problems such as inability to achieve oral administration, achieve cost control, reduce patient burden, and simple operation Effect
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
preparation example Construction
[0048] The preparation method of the vorinostat derivative based on palladium carbon of the present invention can refer to as follows:
[0049] Condensation reaction with vorinostat, followed by deprotection and reduction reaction to obtain the vorinostat derivative;
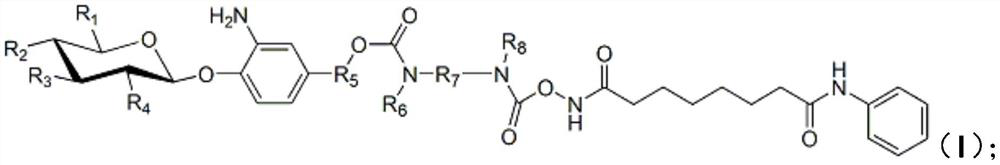
[0050] In formula (i), R 9 -R 12 are independently hydrogen, protected hydroxyl, or protected carboxyl, wherein, R 9 -R 12 There is at least one protected hydroxyl group or protected carboxyl group; preferably, R 9 -R 12 are independently protected hydroxyl or protected carboxyl; more preferably, R 9 -R 12 There is at least one protected hydroxyl group and one protected carboxyl group in, for example, R 9 is a protected carboxyl group, R 10 -R 12 is a protected hydroxyl group; alternatively, R 12 is a protected carboxyl group, R 9 -R 11 is a protected hydroxyl group; or, it can be R 9 is a protected hydroxyl group, R 10 -R 12 is a protected carboxyl group; or, R 12 is a protected hydroxyl group...
Embodiment 1
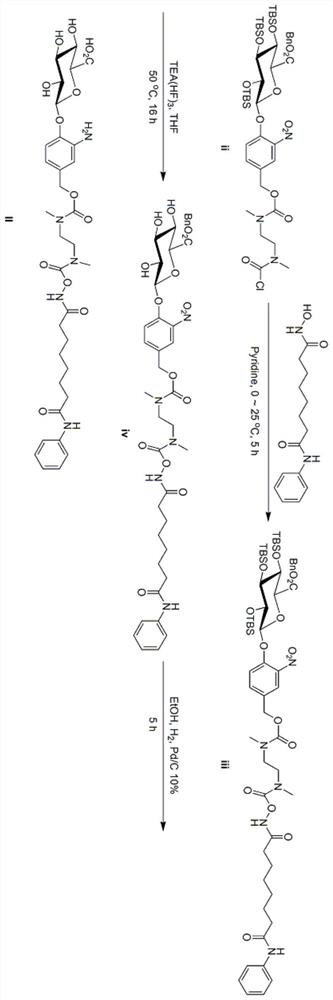
[0075] Example 1 O-[NN-dimethyl-N-4-(2,3,4-tri-O-tert-butyldimethylsilyl-6-benzyl-β-D-glucopyranose Preparation of acid-1-yl)-3-nitrobenzyloxycarbonylethylenediamine]-formyl-vorinostat (iii)
[0076] Dissolve 913 mg (3.46 mmol) of vorinostat in 20 mL of pyridine, and cool at 0° C. for 5 minutes. At 0°C, 3 g (3.14 mmol) of compound (ii) was added to the above solution to form a red suspension. The suspension was warmed up to 25 °C, kept stirring for 5 hours, and then 100 mL of water was added to quench the reaction. The reaction solution was extracted with ethyl acetate (50mL×3 times), the combined organic layers were sequentially extracted with 0.5M HCl (30mL×4 times), saturated brine (30mL×2 times), dried over anhydrous sodium sulfate, and evaporated by rotary evaporation concentrated, and separated by preparative chromatography (dichloromethane:methanol=50:1) to obtain 3.4g of intermediate iii with a yield of 92%.
[0077] Characterization of the product: 1 HNMR (400MHz,...
Embodiment 2
[0078] Example 2 O-[NN-dimethyl-N-4-(6-benzyl-β-D-pyranoglucuronic acid-1-yl)-3-nitrobenzyloxycarbonylethylenediamine]- Preparation of formyl-vorinostat (iv)
[0079] Dissolve 3.4g (2.87mmol) of intermediate iii in 18mL tetrahydrofuran, add 4.63g (28.7mmol) of triethylamine trihydrofluoride at 25°C, heat to 50°C, and keep the reaction for 16 hours. After the reaction is complete, the reaction solution is concentrated with a rotary evaporator, 50 mL of water is added to the concentrated solution, extracted with ethyl acetate / acetonitrile (3 / 1) (20 mL×4 times), the organic layers are combined, washed twice with saturated saline, and anhydrous sulfuric acid Sodium-dried, filtered, the filtrate was concentrated with a rotary evaporator, and separated by preparative chromatography (dichloromethane:methanol=50:1) to obtain 5.0 g of crude product iv with a yield of 74%, which was directly put into the next reaction.
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More