

Method for constructing prokaryote RNA sequencing library
A prokaryotic and sequencing library technology, applied in the field of prokaryotic RNA sequencing library construction, can solve the problems of complex rRNA blocking primer operation, low primer retrieval efficiency and specificity, and low number of retrieved genes, and achieves low preference. , to achieve the effect of preference and avoid degradation
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image


Examples
Embodiment 1
[0048] Example 1 Construction of cultured Escherichia coli RNA sequencing library
[0049] 1. Total RNA extraction.
[0050] Bacteria were collected by centrifugation. Bacterial total RNA was extracted using Tiangen culture cell / bacterial total RNA extraction kit (DP430).
[0051] 2. Connect a specific base sequence at the 3' end of bacterial RNA (that is, the extended sequence of this embodiment, such as the nucleotide sequence shown in SEQ ID No. 3)
[0052] 2.1 The ligation reaction solution contains: 1×T4 RNA Ligase Buffer, 0.5 μM 3'-end RNA extension sequence (SEQID No. 3: 5'- / 5rApp / NNNNNNNNNNNNNNNNCTGTCTCTTATACACATCTGACGCTGCCGA / 3ddC / -3', 5rApp represents 5'-phosphorylated ribose Nucleotide base A, 3ddC denotes 3'-end blocked deoxyribonucleotide base C, N denotes random base), 15% PEG8000, 1 U / μL RNase inhibitor, 10U / μL T4 RNA Ligase.
[0053] 2.2 Incubate at 4°C for 16 hours. Purify RNA using an RNA purification kit.
[0054] 3. Reverse transcription and cDNA amplif...
Embodiment 2
[0076] Example 2 Sequencing and data analysis of the files obtained in Example 1
[0077] 1. Perform PE100 paired-end sequencing using the Illumina NextSeq platform. Among them, the first 16 bits of the sequencing data read by read1 are molecular tags, and the 16-100 bits are the reverse complementary sequence of the original mRNA; the sequencing data read by read2 is the forward sequence of the original mRNA.
[0078] 2. After reverse complementing the 16-100 digit data of read1, compare the reference genome with the data of read2 at the same time, and count the sequencing results of each gene aligned in the reference genome. The comparison of molecular tags and their corresponding sequencing results was counted, and all the sequencing results with the same molecular tag and compared to the same gene were combined into one expression count. Summarize the expression times of all genes to generate sequencing results.
[0079] 3. Summary of results.
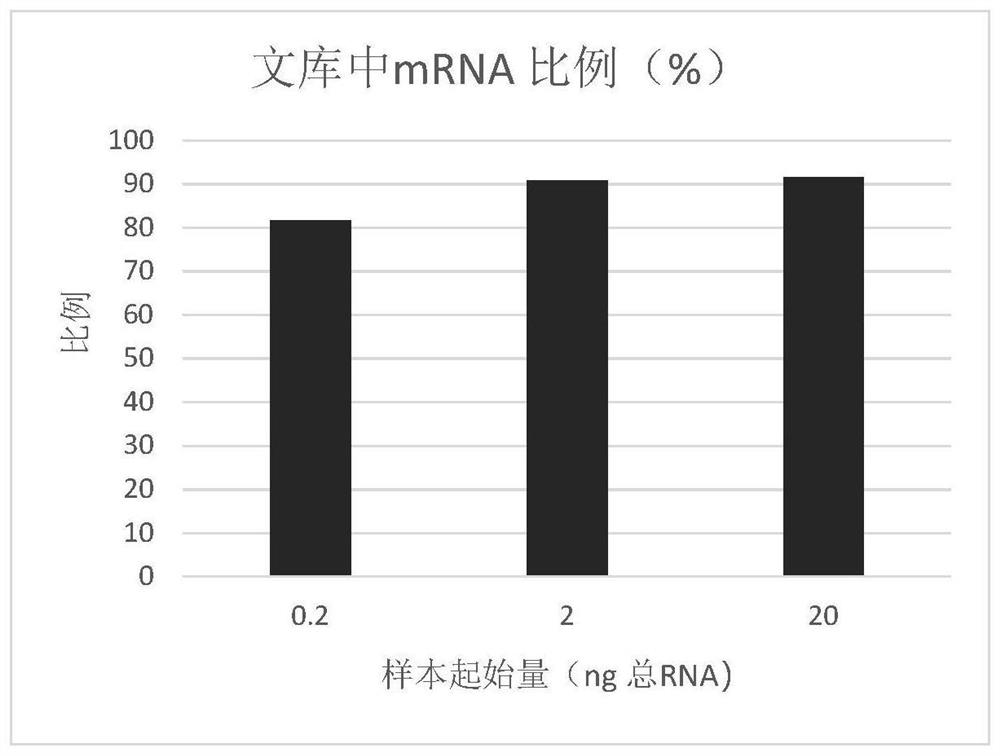
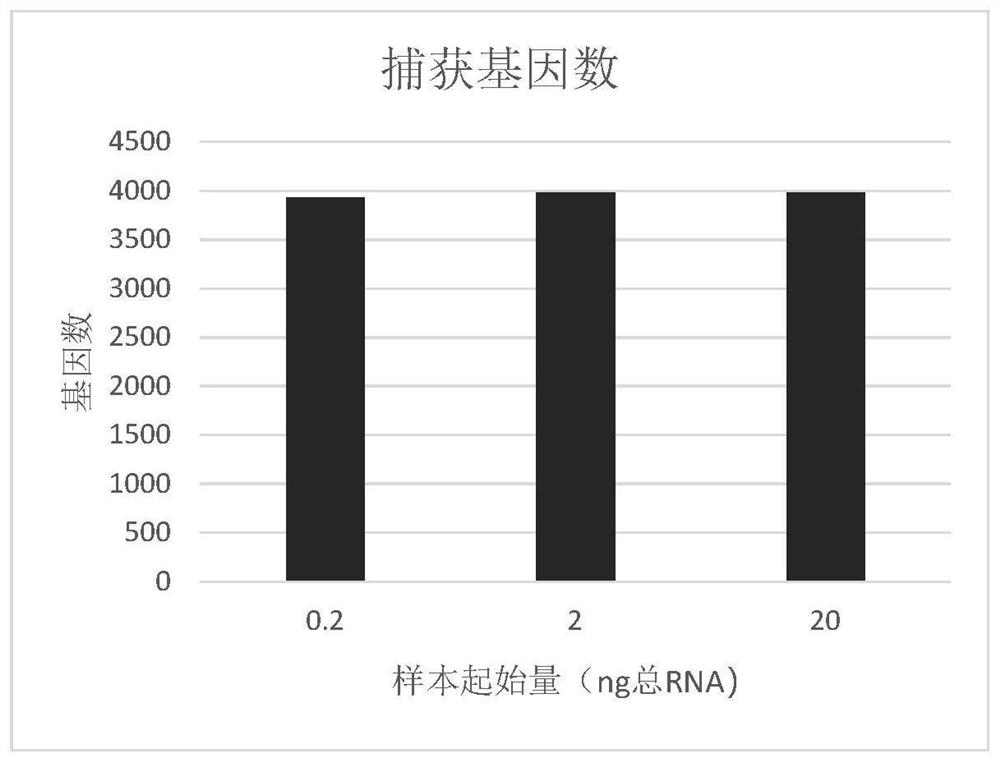
[0080] 3.1 The ratio of ...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More