

Conversion of hydroxy group in certain alcohols into fluorosulfonate ester or trifluoromethylsulfonate ester
A technology of perfluoroalkyl sulfonate and fluorosulfonate, which is applied in the preparation of sulfonate ester, organic chemical method, sulfuric acid ester preparation, etc., can solve the problem of difficult preparation of acid anhydride, toxicity, and high cost of trifluoromethanesulfonic acid And other issues
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
Embodiment 3
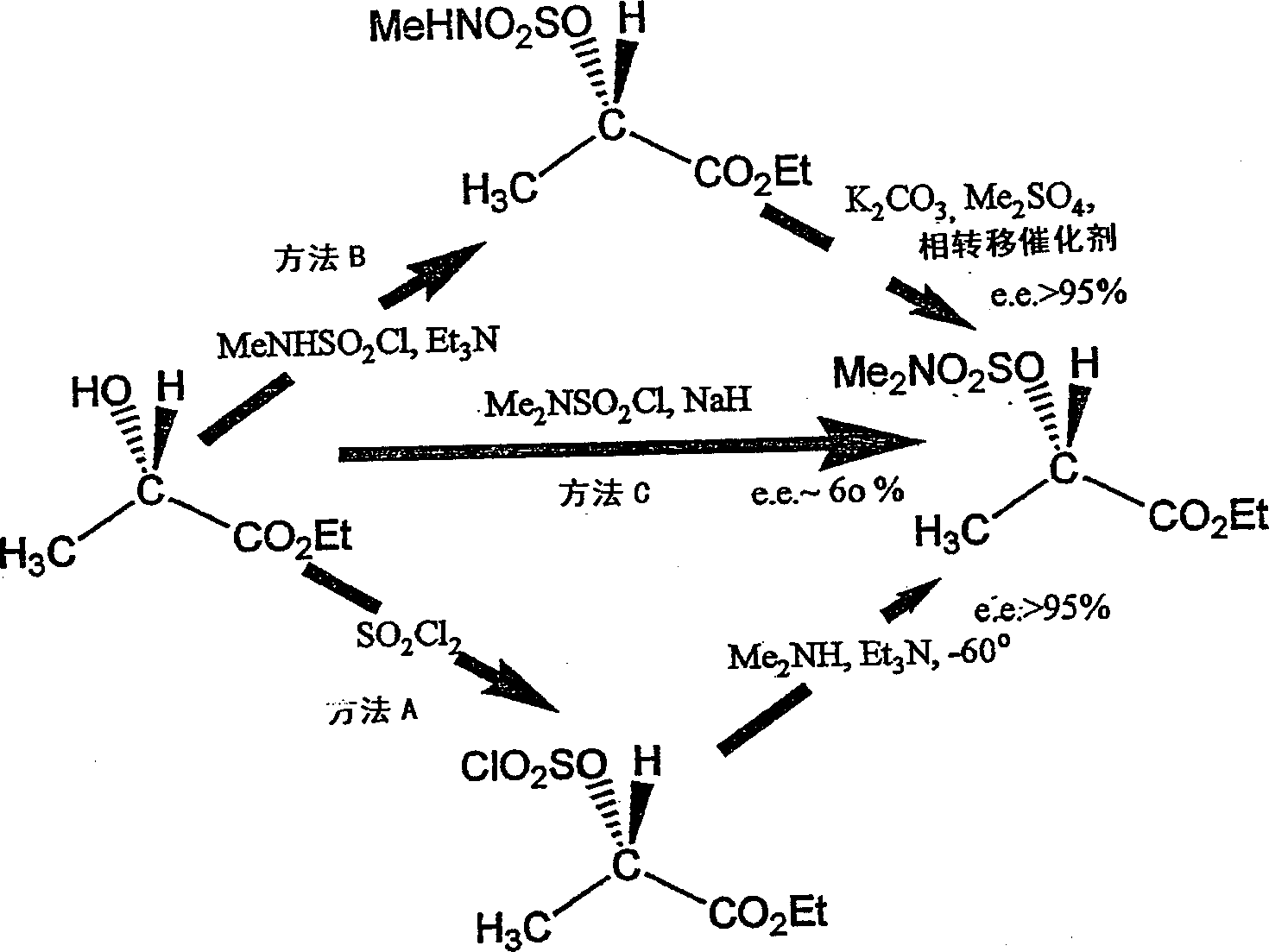
[0038] Note: N-methylsulfamoyl chloride and N,N-dimethyl analogs described in the following examples are given by G.Weiss and G.Schulze, Liebig's Ann.Chem.1969,729,40, General method preparation, productive rate is more than 95%, and this method is after adding the titanium tetrachloride of catalytic amount (1ml), utilizes the water bath of constant temperature control and the gas catcher drawn by condenser, heats corresponding under reflux A suspension of amine hydrochloride (1 g.mol.), sulfonyl chloride (2.1 g.mol.) and acetonitrile (200 ml) for 1.5 times the time necessary for complete dissolution. In the case of the N-methyl compound, this took more than 72 hours, whereas it took less than 8 hours with the N,N-dimethyl chloride (the latter being commercially available). Then, after solvent removal, the product was distilled by bulb-to-bulb method (ice-cooled collector) below 0.5 mmHg (heat bath or oven below 130°C). N-methylsulfamoyl chloride is distilled at 70-75°C / 0.1mm...
Embodiment 4
[0041] Trifluoromethanesulfonic acid (10.88 g, 72 mmol) was added to dry CCl of the sulfamoyloxy compound (16.81 g, 74.71 mmol) prepared in Example 2 under vigorous stirring at 0 °C 4 (40 mL) solution, and stirring was continued at room temperature for 8 hours. The resulting N,N-dimethylsulfamic acid was filtered off and washed with CCl 4 Wash the filtrate with cold 0.5 M NaHCO 3 Wash, dry and remove solvent at room temperature 23 mmHg. Fractional distillation (Teflon column) residue, to obtain triflate (or salt) 12.94g (yield 71.5%), boiling point 32-34 ℃ / 0.07mmHg, n.m.r.1.31 (t, 3H), 1.67 ( d, 3H), 4.28(q, 2H); i.r. 1767, 1428, 1217, 1153, 961, 622; [α] 21 :+43.7 0 .H.H.Paulsen, P.Himpkamp, and T.Peters, Liebigs Ann.Chem.1986,664, Utilize trifluoromethanesulfonic anhydride-pyridine to prepare S-enantiomer, reported boiling point is 40.5°C / 0.9mmHg, [α ]:-40 0 ; while the commercially available S-enantiomer has a reported boiling point of [α]-44 0 ±2. Example 5 Ethyl...
Embodiment 5
[0042] Fluorosulfonic acid (6.1 g, 61 mmol) was added to dry CCl at 0 °C 4 Of the sulfamoyloxy compound prepared in Example 2 (13.55 g, 60.22 mmol) in (35 mL), the whole mixture was stirred vigorously at room temperature for 7 hours. Treat according to Example 4, and distill (boiling point is 34-36 ℃ / 0.1mmHg) to obtain product, yield is 9.23g (82%), n.m.r.1.31(t, 3H), 1.56(d, 3H), 4.25(t, 2H),5.19(q,1H), 19 Fn.m.r.: singlet at 98.96 p.p.m; i.r.1760.5, 1447, 1224, 980, 839; [α] 16 :+44.34 0 , there is no high-resolution [M + ]peak. Conversion experiment example 6 Preparation of ethyl S-2-acetoxypropionate from R-trifluoromethylsulfonate (or salt)
PUM
| Property | Measurement | Unit |
|---|---|---|
| melting point | aaaaa | aaaaa |
Abstract
Description
Claims
Application Information
 Login to View More
Login to View More