

Preparation method of lurasidone
A technology of lurasidone and diketone, applied in the field of preparation of pharmaceutical compounds, can solve the problems of unstable acid anhydride, low process yield and the like, and achieve the effects of simple operation, simple purification and convenient recovery
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

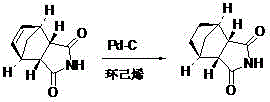
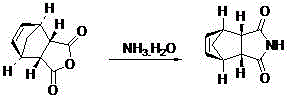
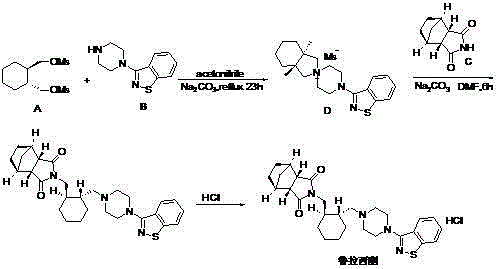
Examples
Embodiment 1
[0025]
[0026] Put 3.8g (17.4mmol) 3-(1-piperazinyl)-1,2-benzisothiazole into 150ml toluene, stir to dissolve, add 5.7g (19.0mmol) (1R,2R)-1,2- Bis(methanesulfonate oxymethyl)cyclohexane, and 7.2g (52mmol) potassium carbonate, heat up, reflux at 110-130°C for 30h, until TLC detects the intermediate 3-(1-piperazinyl)-1,2 - Benzisothiazole basically disappears (main peak LC-MS: M + =328.2), add 3.1g (18.7mmol) (3αR,4S,7R,7αS) 4,7-methylene-1H-isoindole-1,3(2H)-dione, stir and heat up, 110-130 Reflux at ℃ for 8 hours, cool, recover toluene under reduced pressure, add 200ml of ethyl acetate to the residue, stir to dissolve, wash with 5% hydrochloric acid solution three times, separate the organic layer, add anhydrous magnesium sulfate to dry for 2 hours, and filter out the desiccant to obtain ethyl acetate solution. The above ethyl acetate solution was evaporated and concentrated to about 80ml, concentrated hydrochloric acid was added dropwise, a large amount of white solid ...
Embodiment 2
[0028] Put 3.8g (17.3mmol) 3-(1-piperazinyl)-1,2-benzisothiazole into 150ml toluene, stir to dissolve, add 7.84g (26.10mmol) (1R,2R)-1,2- Bis(methanesulfonate oxymethyl)cyclohexane, and 7.2g (52mmol) potassium carbonate, heat up, reflux at 110-130°C for 24h, until TLC detects the intermediate 3-(1-piperazinyl)-1,2 - Benzisothiazole basically disappears, add 4.3g (26.1mmol) (3αR,4S,7R,7αS) 4,7-methylene-1H-isoindole-1,3(2H)-dione, stir and heat up , reflux at 110-130°C for 4 hours, cool, recover toluene under reduced pressure, add 200ml of ethyl acetate to the residue, stir to dissolve, wash with 5% hydrochloric acid solution three times, separate the organic layer, add anhydrous magnesium sulfate to dry for 2 hours, filter off Desiccant, in ethyl acetate solution. The above ethyl acetate solution was evaporated and concentrated to about 80ml, concentrated hydrochloric acid was added dropwise, a large amount of white solid was precipitated, and filtered by suction to obtain 6....
Embodiment 3
[0030]Put 3.8g (17.3mmol) 3-(1-piperazinyl)-1,2-benzisothiazole into 120ml toluene, stir to dissolve, add 4.2g (13.8mmol) (1R,2R)-1,2- Bis(methanesulfonate oxymethyl)cyclohexane, and 7.2g (52mmol) potassium carbonate, heat up, reflux at 110-130°C for 30h, until TLC detects the intermediate 3-(1-piperazinyl)-1,2 - Benzisothiazole basically disappears, add 2.8g (13.9mmol) (3αR,4S,7R,7αS) 4,7-methylene-1H-isoindole-1,3(2H)-dione, stir and heat up , reflux at 110-130°C for 8 hours, cool, recover toluene under reduced pressure, add 150ml of ethyl acetate to the residue, stir to dissolve, wash with 5% hydrochloric acid solution three times, separate the organic layer, add anhydrous magnesium sulfate to dry for 2 hours, filter off Desiccant, in ethyl acetate solution. The above ethyl acetate solution was evaporated and concentrated to about 60ml, concentrated hydrochloric acid was added dropwise, a large amount of white solid was precipitated, and filtered by suction to obtain 5.24g...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More