

A Method for Analyzing High-Throughput Sequencing Gene Expression Levels Using Text Alignment
A gene expression level, high-throughput technology, applied in the field of bioinformatics, can solve problems such as large differences and differences in results, and achieve the effect of reducing workload, simple method, and simple and fast splicing
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image



Examples
Embodiment 1
[0062] Example 1 Camellia high-throughput sequencing
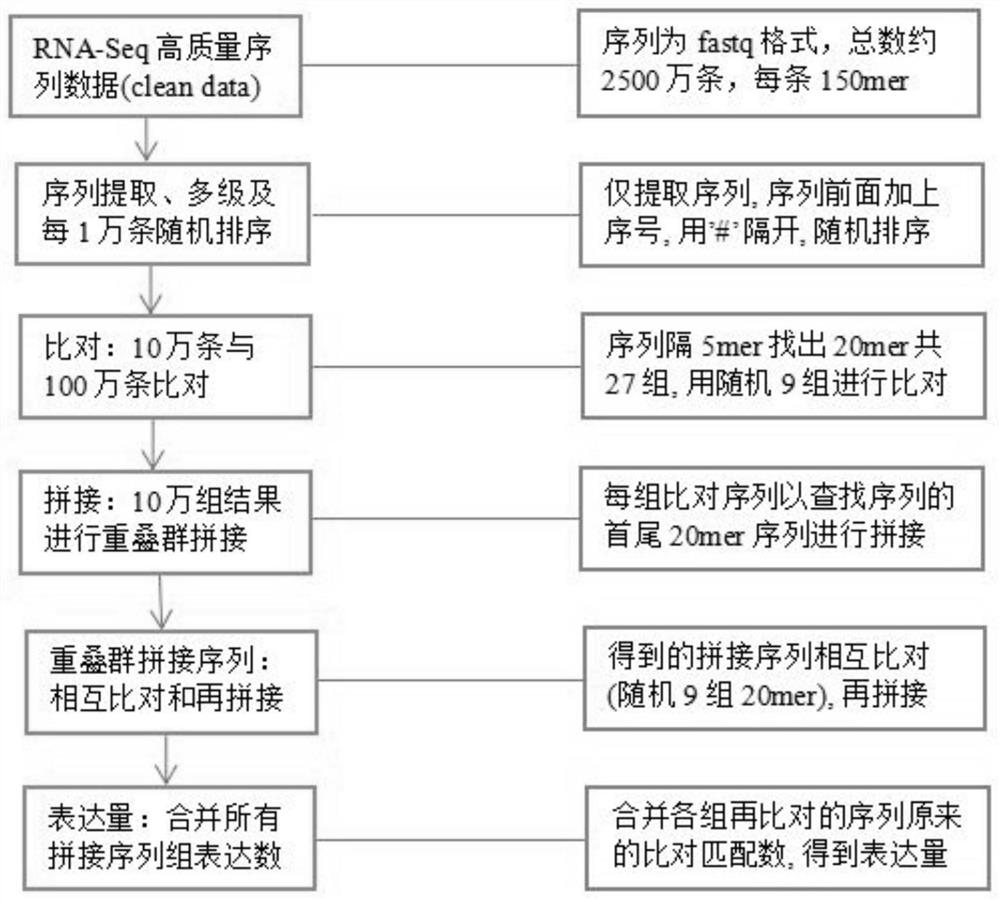
[0063] The company provides sequencing services for the fully developed leaves and petals of unopened buds of camellia flowering branches during the flowering period, including total RNA extraction and library construction, paired-end sequencing (Paired-End, Illumina HiSeq 4000). The sequence format is fastq, submit 6G high-quality data (clean data) and 7G unprocessed raw sequencing data (raw data), each sequence length is 150mer, and merge the double-ends to obtain about 50 million sequencing sequences for each sample.
Embodiment 2
[0064] Example 2 Sequencing sequences are numbered, broken up, and randomly combined
[0065] Extract the high-throughput sequencing sequences obtained in Implementation 1, and only keep the sequences. Each sequence is numbered. There are 50 million sequences in this sequence (about 25 million sequences are generated by paired-end sequencing respectively), and the sequencing of one end is from the first sequence. The serial numbers from the sequence to the 25 millionth sequence are 00000001-25000000. Then use step-by-step random sorting every 100,000 → 1 million → 50,000, and merge the sequences in a random way, cut the sequence documents, sort them randomly, and merge all the sequences into one document. Among them, each 1 million sequence documents cut according to 1 million pieces are divided into several directories, and then randomly sorted every 10,000 pieces and then randomly merged to obtain 1 million sequences, and the documents obtained from all directories are rando...
Embodiment 3
[0066] Example 3 Among 1 million sequences, 100,000 are selected as query sequences for comparison, and the expression level of each query sequence is obtained
[0067] Randomly select the 1,000,000 fragmented sequences in Example 2 of the above steps, divide the 1,000,000 sequences into every 100,000 sequences, and randomly select one 100,000 sequences as the query sequence.
[0068] In the above 100,000 query sequences, for each sequence, perform the following operations:
[0069] 1. Take 20 consecutive nucleotide sequences (20mer) every 5 nucleotides, and each query sequence can be divided into 27x20mer and short sequences;
[0070] 2. In each query sequence, randomly select 9 short sequences of 20mer;
[0071] 3. At least 9 randomly selected short sequences of 20mer are used to match and compare with 1 million sequences. At the same time, the complementary strands of at least 9 20mer short sequences are also matched and compared with 1 million sequences, and the matching ...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More