

A kind of preparation method of mesoporous double-layer organosilicon sphere
A silicone and double-layer technology, applied in the preparation of organic compounds, medical preparations of non-active ingredients, organic chemistry, etc., can solve the problem that it cannot be used as a drug release carrier of different sizes, affects the function of silicon spheres, and cannot be completely removed Templating agent and other issues
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

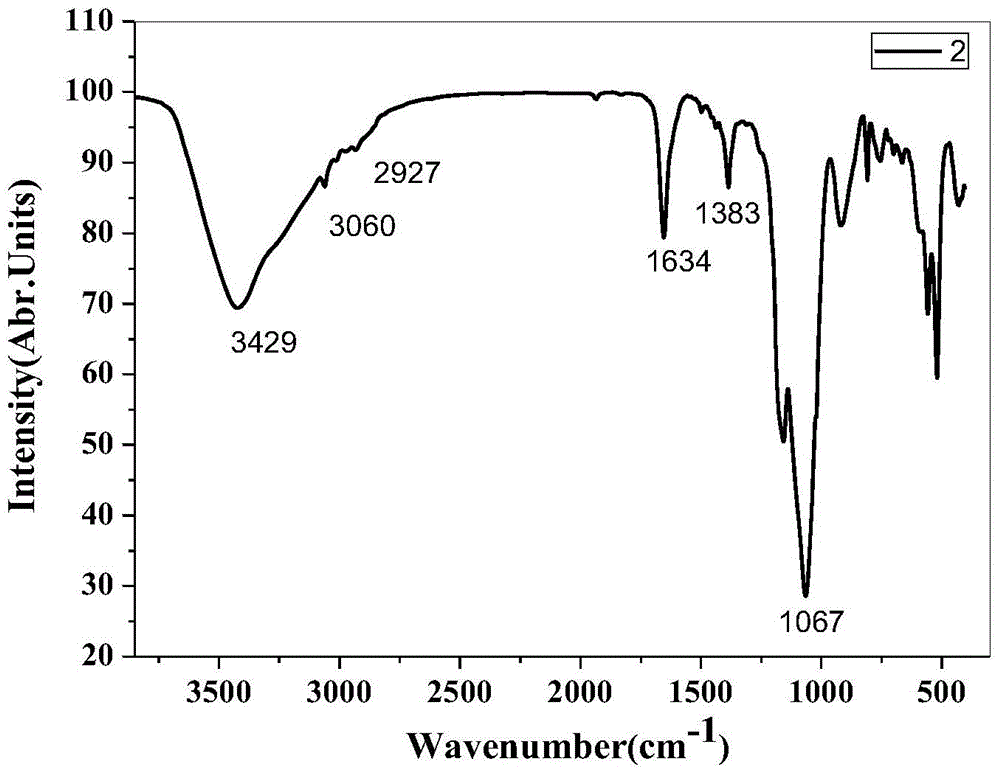
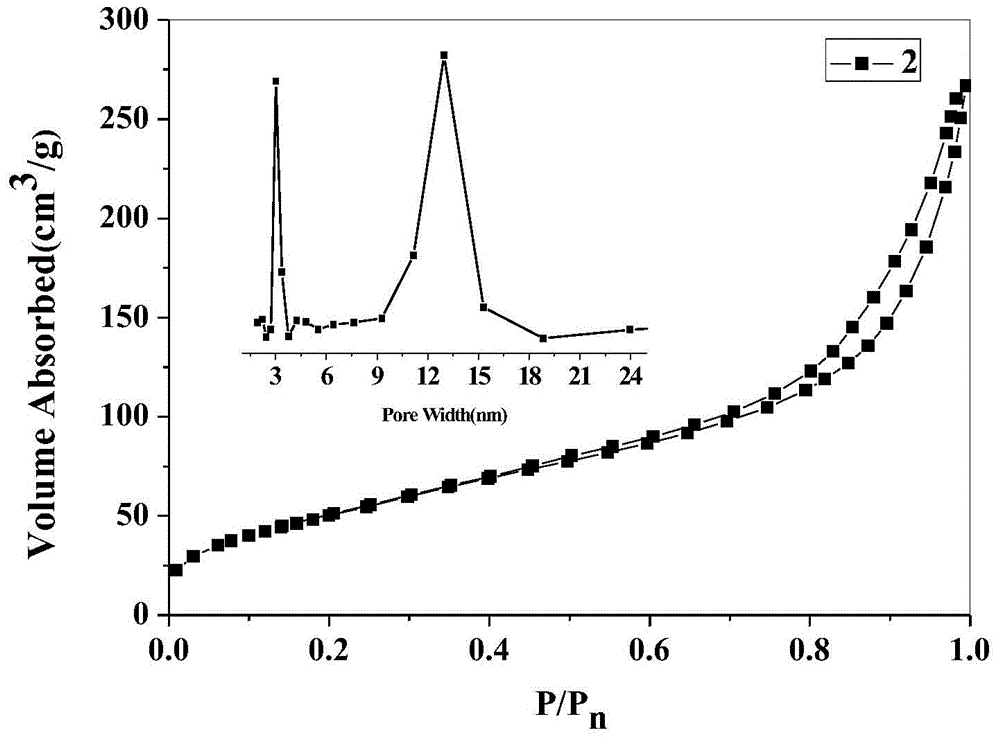

Examples
preparation example Construction
[0048] A preparation method of mesoporous double-layer silicone spheres, comprising the following steps:
[0049] 1) Tetraethylammonium bromide, Al(OH) 3 and Ca(OH) 2 Add in deionized water and mix evenly to obtain a mixed solution A. Ultrasonic the mixed solution A for 5 to 24 hours until it is completely uniform, then take it out, then heat it to 80-120°C and stir evenly to obtain a mixed solution B; ,
[0050] The tetraethylammonium bromide, Al(OH) 3 , Ca(OH) 2 And the molar ratio of deionized water is 0.5~0.8:0.3~0.5:0.5:3000~4000;
[0051] 2) Add ammonia water to the mixed solution B obtained in step 1), and then add 1,4-bis(triethoxysilyl)benzene dropwise at a rate of 10 drops per minute and adjust the pH to 10-13, The temperature rises to 80-100°C, and after reacting for 5-7 hours, a mixed solution C is obtained; wherein,
[0052] The molar ratio of tetraethylammonium bromide, ammonia water and 1,4-bis(triethoxysilyl)benzene is 0.5-0.8:0.01-0.03:1;
[0053] 3) Tr...
Embodiment 1
[0062] At room temperature, 0.38gTAB (tetraethylammonium bromide), 0.1gAl(OH) 3 with 0.135gCa(OH) 2 Add 150 mL of deionized water and mix evenly (mol ratio of Ca / Si=0.5). The whole system was ultrasonicated for 6 hours, and after the system was completely uniform, it was taken out, heated to 100°C and stirred evenly. Join NH 3 .H 2 After 0.01 mL, 1.0 g (2.5 mmol) of 1,4-Bis(triethoxysilyl)-benzene (1,4-bis(triethoxysilyl)benzene) was slowly added dropwise (pH=12.8). After reacting at 100°C for 5 hours, the reaction liquid was transferred to a high-pressure reactor and reacted at 110°C for 72 hours, and the solid was obtained by suction filtration. The solid was extracted with HCl-EtOH at 80 °C for 12 h. The reaction product was dried in a vacuum oven at 60° C. for 5 hours. This gave 0.82 g of a white solid.
[0063] 1.0g of compound 1 and 0.1g of Al(OH) 3 Add 5mL triethanolamine, add 0.12g CTAB and 0.08g CTAC in 100mL water once, stir for 1h, the temperature rises to 6...
Embodiment 2
[0065] At room temperature, 0.21gTAB (tetraethylammonium bromide), 0.09gAl(OH) 3 with 0.08gCa(OH) 2 Add 140 mL of deionized water and mix evenly (mol ratio of Ca / Si=0.5). The whole system was ultrasonicated for 12 hours. After the system was completely uniform, it was taken out, heated to 90°C and stirred evenly. Join NH 3 .H 2 After 0.02 mL, 0.8 g (2 mmol) of 1,4-Bis(triethoxysilyl)-benzene (1,4-bis(triethoxysilyl)benzene) was slowly added dropwise (pH=11.8). After reacting at 80° C. for 8 h, the reaction liquid was transferred to a high-pressure reactor to react at 100° C. for 3 days, and the solid was obtained by suction filtration. The solid was extracted with HCl-EtOH at 60 °C for 24 h. The product was dried in a vacuum oven at 60°C for 5 hours. This gave 0.42 g of a white solid.
[0066] 0.7g of compound 1 and 0.05g of Al(OH) 3 Add 3mL of triethanolamine, add 0.07g of CTAB and 0.04g of CTAC in 80mL of water once, stir for 1 hour, the temperature rises to 80°C, di...
PUM
| Property | Measurement | Unit |
|---|---|---|
| pore size | aaaaa | aaaaa |
Abstract
Description
Claims
Application Information
 Login to View More
Login to View More