

Method for removing human gene sequence in macro genome sequencing data
A technology of sequencing data and metagenomics, which is applied in the field of genetic engineering, can solve the problems of insufficient removal of human genome sequences and high false positives in microbial analysis, and achieve the effect of increasing speed and saving computing resources
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
Embodiment Construction
[0062] In order to express the present invention more clearly, the present invention will be further described below in conjunction with the accompanying drawings.
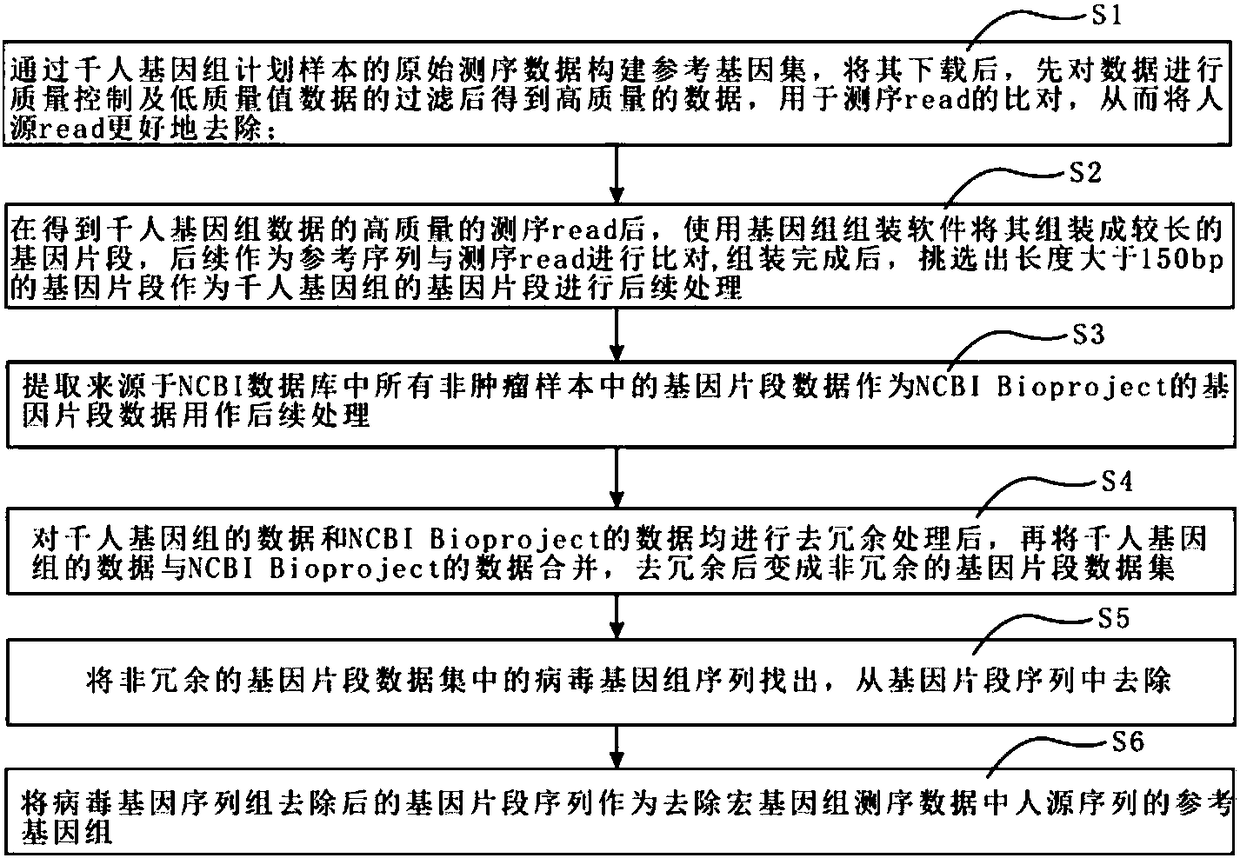
[0063] see figure 1 , the method for removing the human gene sequence in the metagenomic sequencing data of the present invention comprises the following steps:
[0064] Step S1: Construct a reference gene set from the original sequencing data of the 1000 Genomes Project samples. After downloading it, first perform quality control on the data and filter low-quality data to obtain high-quality data for comparison of sequencing reads , so as to better remove human-sourced reads;
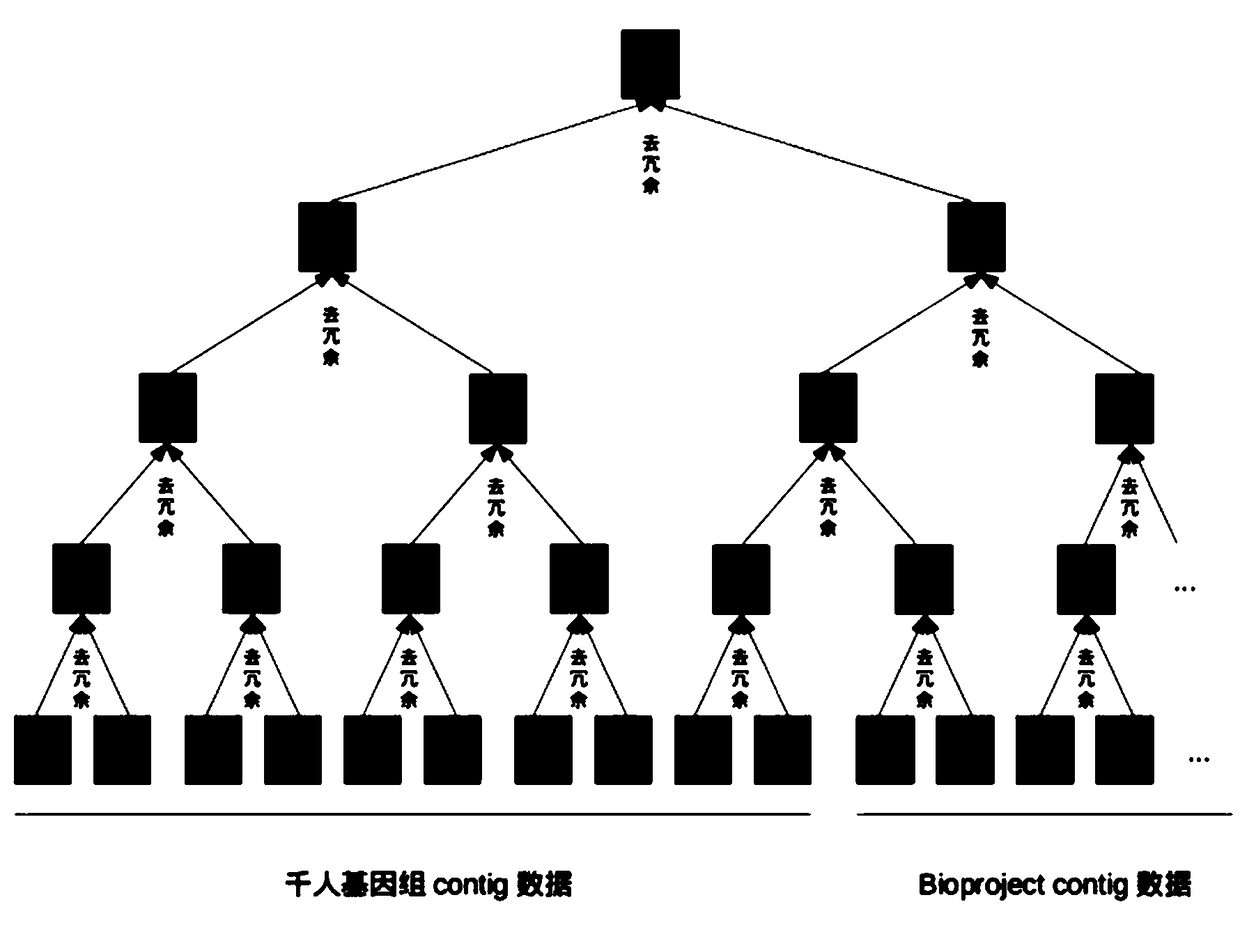
[0065] Step S2, after obtaining the high-quality sequencing reads of the Thousand Genomes data, use genome assembly software to assemble them into longer gene fragments, and then compare them with the sequencing reads as a reference sequence. The 150bp gene fragment is used as the gene fragment of the Thousand Human Genome for subsequent...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More