

Methods for dynamic vector assembly of DNA cloning vector plasmids
a plasmid and vector technology, applied in the field of cloning vector plasmids, can solve the problems of many sites of 6 or less nucleotides being useless, difficult to manipulate at best, statistically very likely,
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Benefits of technology
Problems solved by technology
Method used
Image

Examples
example 1
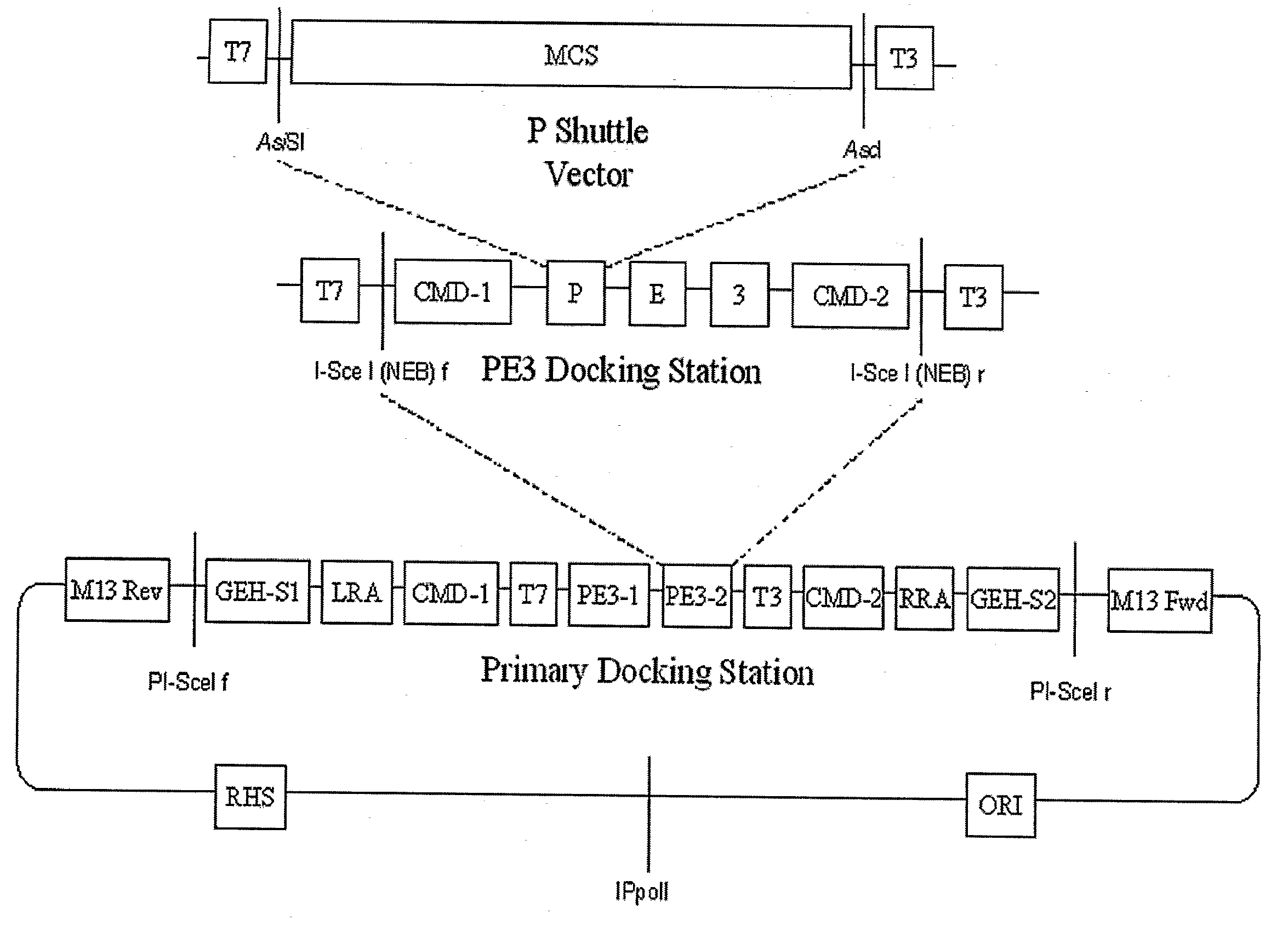
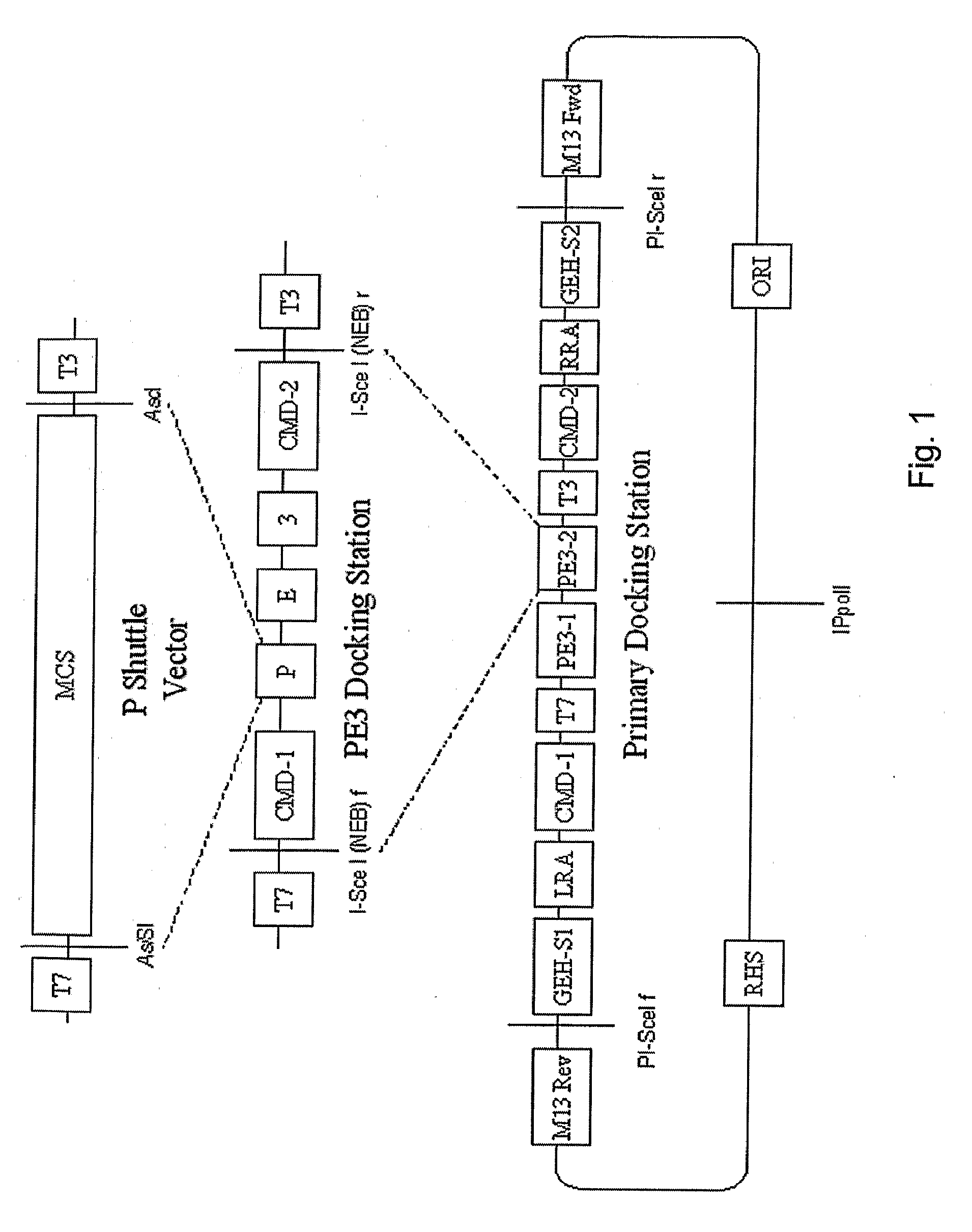
PE3 Docking Plasmid
[0180]As an example of the method of practicing the present invention, a transgene can be constructed containing these elements:
[0181]1. Nucleotide sequences of the human promoter for surfactant protein C(SP-C);
[0182]2. Sequences encoding the protein product of the mouse gene granulocyte-macrophage colony-stimulating factor-receptor beta c (GMRβc);
[0183]3. Rabbit betaglobin intron sequences; and
[0184]4. SV40 poly-A signal.
[0185]The SP-C sequences contain internal BamH1 sites, and can be released from its parental plasmid only with Not1 and EcoR1. GMRβc has an internal Not1 site, and can be cut from its parental plasmid with BamH1 and Xho1. The rabbit betaglobin intron sequences can be cut out of its parental plasmid with EcoR1. The SV-40 poly-A tail can be cut from its parental plasmid with XhoI and Sac1. Because of redundancy of several of endonuclease sites, none of the parental plasmids can be used to assemble all the needed fragments.
[0186]The steps used to bu...
example 2
Dynamic Vector Assembly
[0192]Dynamic Vector Assembly is illustrated in the following example:
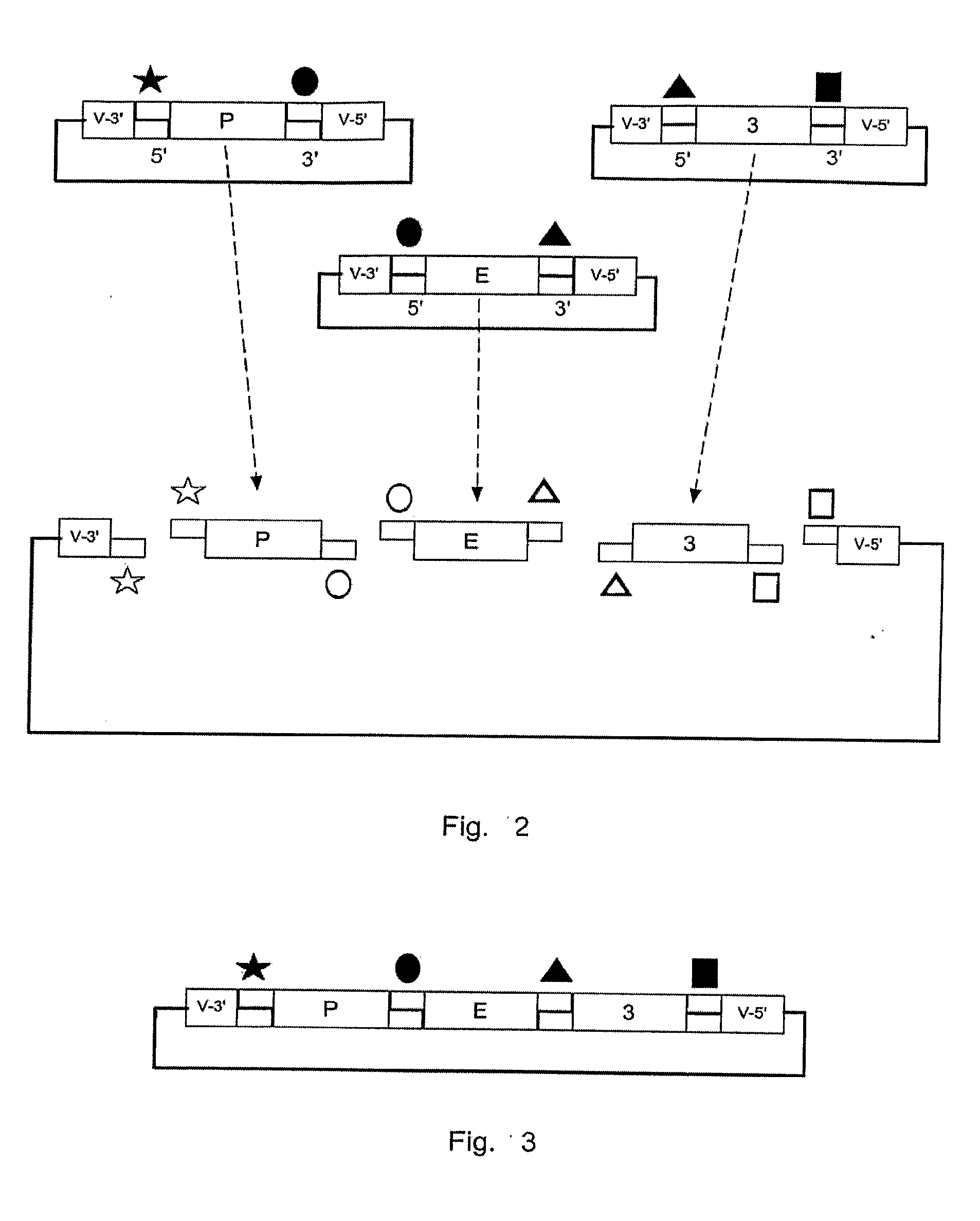
[0193]1. Promoter sequences from the human cytomegalovirus (CMV) are inserted into a P Shuttle Vector (SVP), having AsiSI and Ase I endonuclease at the 5′ and 3′ portions, respectively. Plasmids are amplified, and the promoter module is cleaved from the vector by AsiS I and Asc I endonuclease digestion and isolated.
[0194]2. Sequences encoding a luciferase protein are inserted into an Expression Shuttle Vector (SVE), having Asc I and Not I endonuclease at the 5′ and 3′ portions, respectively. Plasmids are amplified, and the Expression module is cleaved from the vector by Asc I and Not I endonuclease digestion and isolated.
[0195]3. Sequences encoding a mammalian intron and SV40 poly-adenylation site are inserted into a 3′ Regulatory Shuttle vector (SV3), having Not I and BsiW I endonuclease at the 5′ and 3′ portions, respectively. Plasmids are amplified, and the Regulatory module is cleaved fr...
example 3
Redesign of a Dynamic Vector Assembly
[0198]If the researcher now wishes to refine the expression pattern so that luciferase is expressed only in a particular tissue or cell-type, he or she can quickly and easily replace the CMV promoter with one that will provide a restricted expression pattern. The following example illustrates the use of the invention to facilitate rapid redesign of pCMV-luc-pA:
[0199]1. A neuron-specific promoter, Neuron-Specific Enolase (NSE), is inserted into a P Shuttle Vector (SVP) and prepared as the Promoter Module in the previous example.
[0200]2. pCMV-luc-pA is cleaved with AsiS I and Asc I to remove the CMV Promoter Module. The remainder of the Docking Vector Plasmid containing intact Expression and Regulatory Modules is isolated.
[0201]3. The NSE Promoter Module is placed in a ligation mixture with the remainder of the Docking Vector Plasmid containing intact Expression and Regulatory Modules. Following incubation for 2 hours, the new ligation mixture is u...
PUM
| Property | Measurement | Unit |
|---|---|---|
| volume | aaaaa | aaaaa |
| structure | aaaaa | aaaaa |
| temperature | aaaaa | aaaaa |
Abstract
Description
Claims
Application Information
 Login to View More
Login to View More