

Compositions comprising triterpenoids
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Benefits of technology
Problems solved by technology
Method used
Image

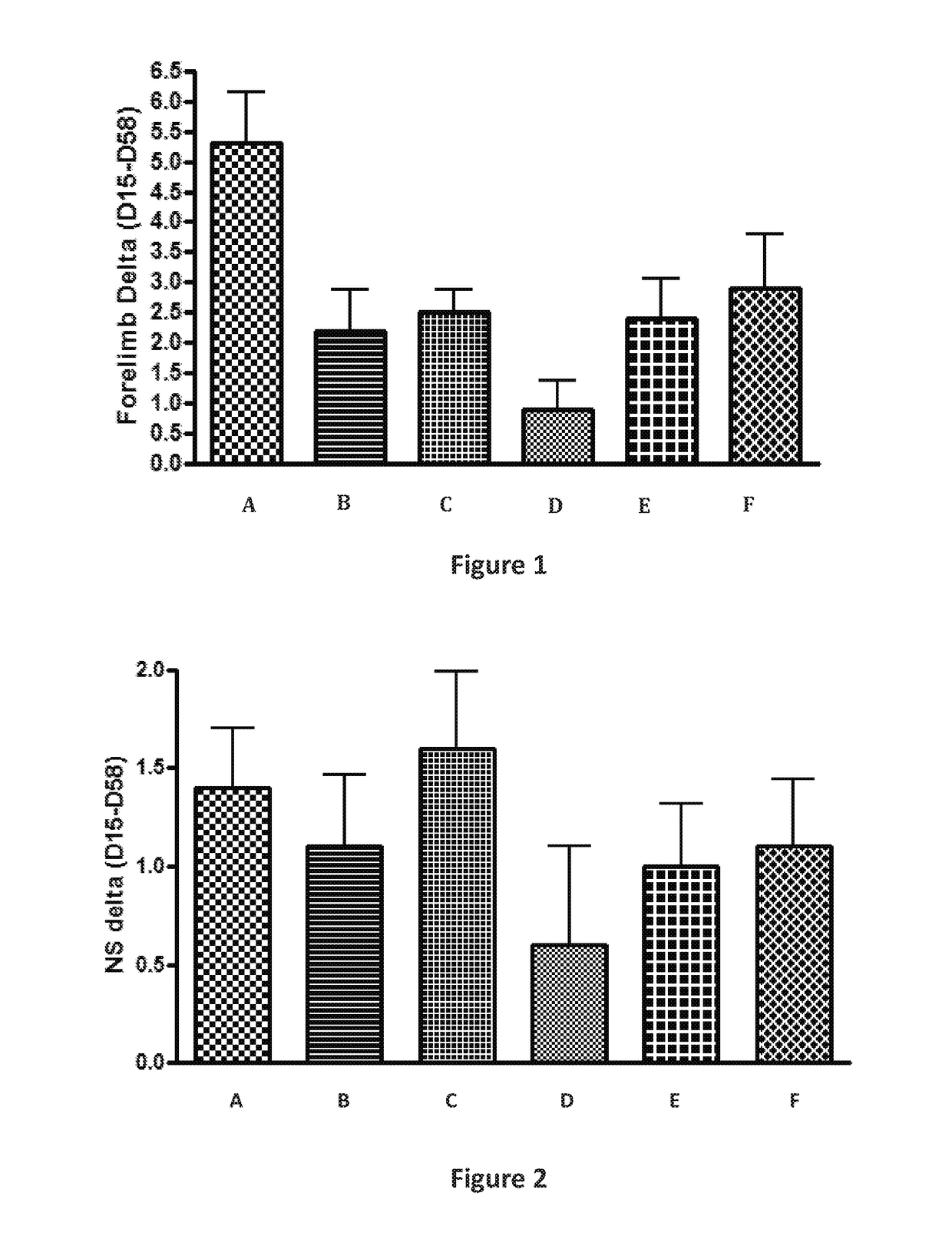
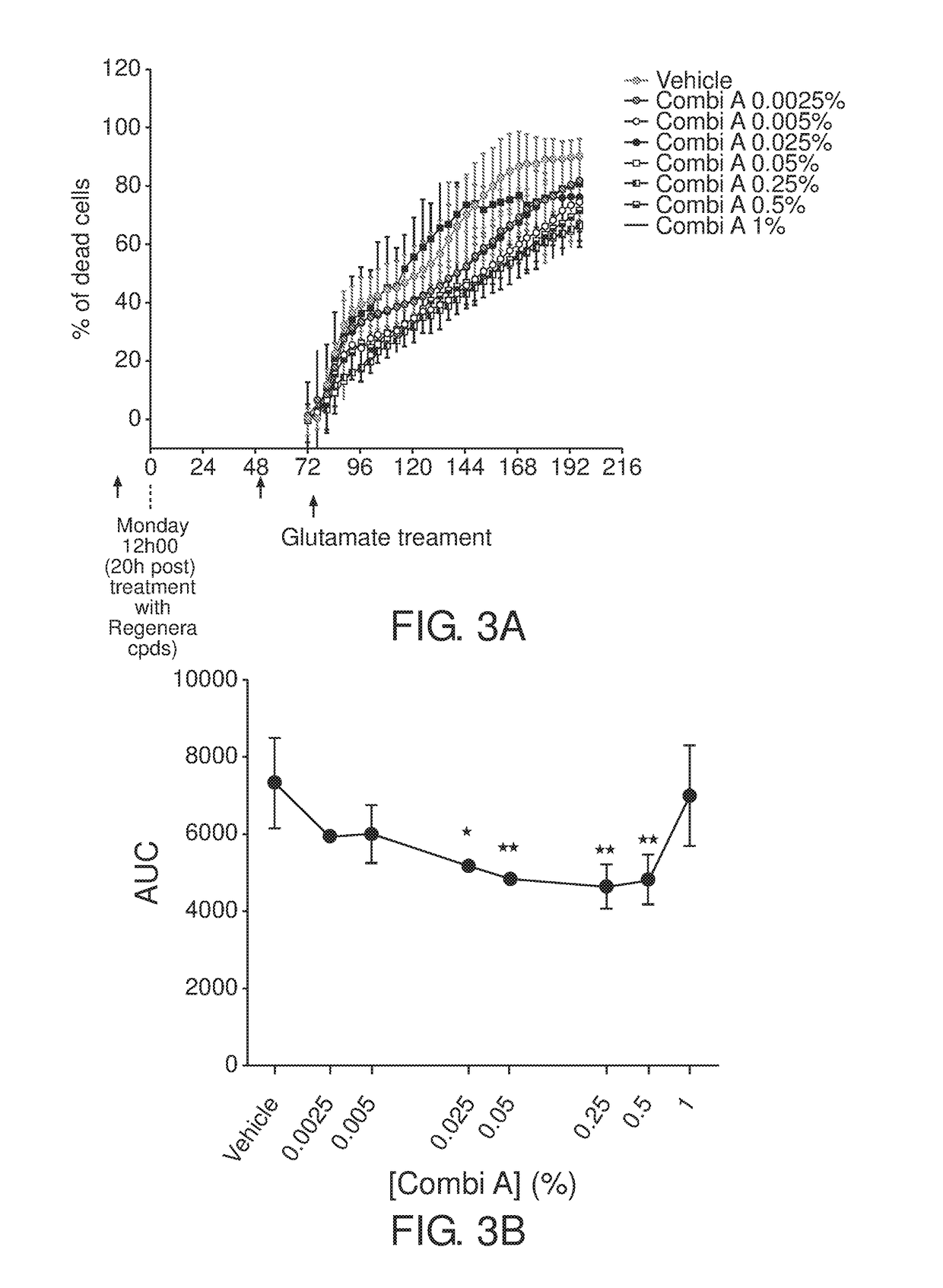
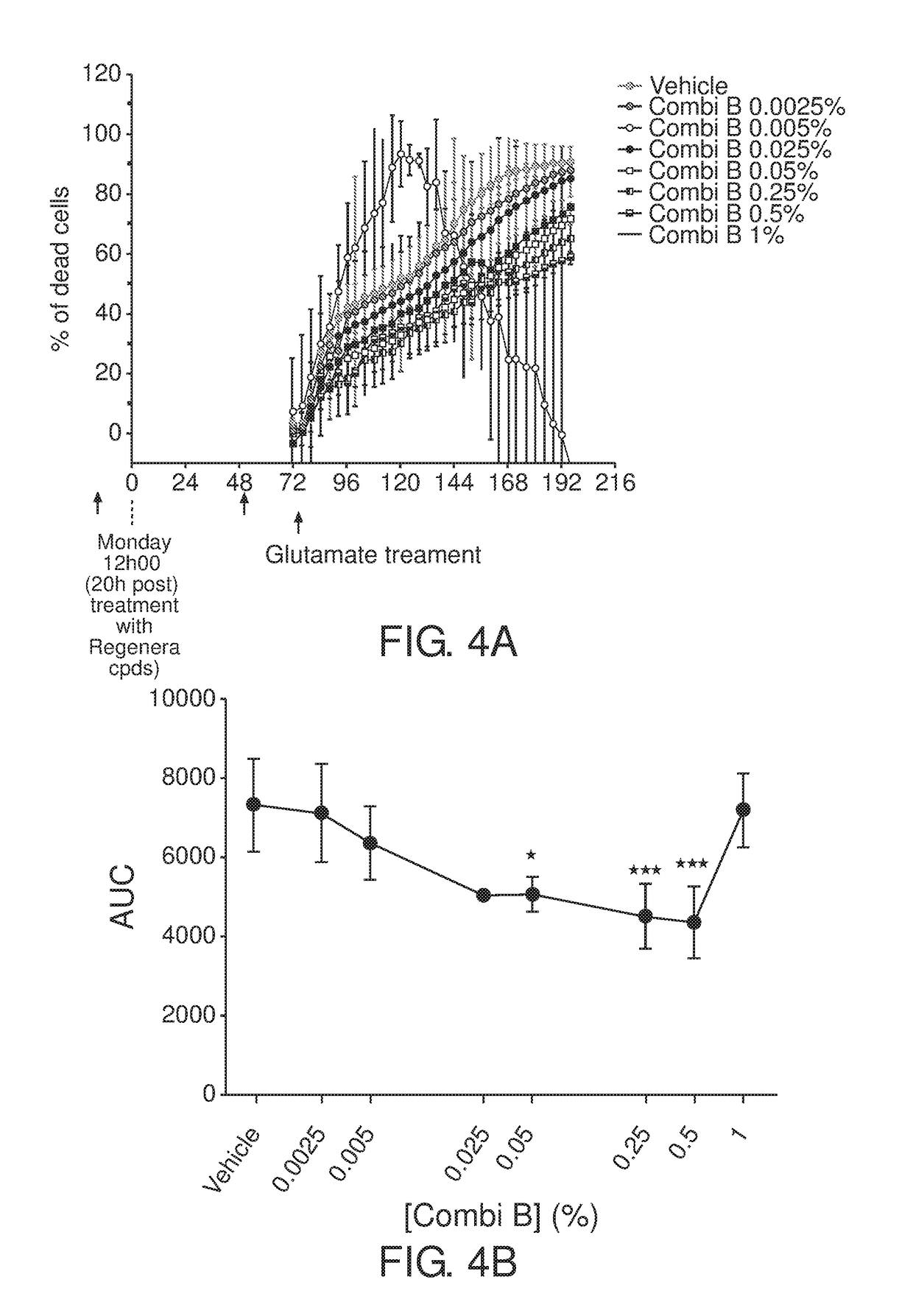
Examples
example 1a
n of Isolated Acidic Fraction of Mastic Gum
[0355]To a 50 gram amount of mastic gum, absolute ethanol (800 ML) was added, and the mixture was left to stand for 24 hours. The mixture was shaken for 30 minutes at 150 rpm and left to stand for two hours. The obtained ethanol solution was decanted from insoluble material into a 3 L round bottom flask. To the insoluble material 400 ML of fresh ethanol was added and the mixture was shaken again 30 minutes at 150 rpm and was left to stand for 30 minutes. The obtained ethanol solution was decanted and added to the first ethanol solution. This step was repeated once more using 200 ML absolute ethanol. This provided 1.4 L of ethanol solution. The ethanol was evaporated using a rotary evaporator, and n-hexane (1.2 Liter) was added to the remaining material, and the mixture was shaken at 150 rpm for 4 hours. It was then left to stand for 4 hours and the hexane solution was decanted from insoluble material into a 3 L Erlenmeyer. To the remaining ...
example 1b
of the Neutral Fraction of Mastic Gum
[0358]The diethyl ether layer Nr.I obtained in Example 1A was transferred to a clean separatory funnel and washed with water (200 ML) and brine (150 ML). It was then dried over anhydrous sodium sulfate. The sodium sulfate was removed by filtration and the diethyl ether was evaporated using a rotary evaporator. This gave about 15 grams of isolated neutral fraction as a white to off-white sticky solid (which will become a very viscous liquid above 35-40° C.). This is about 50% yield based on the extract obtained after the ethanol / hexane extraction presented in Example 1A. This particular isolated neutral fraction obtained from mastic gum as described here is termed “Neutral Mixture 1”. Based on the starting 50 grams of Mastic gum, the yield for this neutral fraction (“Neutral Mixture 1”) is about 30%. Typical yields of this neutral fraction from mastic gum range from about 25 to about 35%.
[0359]The mass-balance of this particular acid-base extracti...
example 2
of a Triterpenoic Acid and Some Neutral Triterpenoids
Synthesis A: Preparation of Oleanonic Acid
[0362]Oleanonic acid was obtained in three steps from oleanolic acid.
[0363]Oleanolic acid was first converted to the corresponding methyl ester by treatment with methyl iodide and potassium carbonate in dimethylformamide (DMF). Oxidation of oleanolic acid methyl ester to oleanonic acid methyl ester was performed using Dess-Martin periodane reagentin dichloromethane (DCM). Hydrolysis of oleanonic acid methyl ester with lithium hydroxide in aqueous THF gave upon acidification the desired oleanonic acid.
Oleanonic Acid Methyl Ester:
[0364]Oleanolic acid (3.66 g, 1.0 eq) was dissolved in DMF (20.0 vol.). K2CO3 (3.3 g, 3.0 eq) was added and mixture was stirred for 10 minutes, then methyl iodide (0.75 ml, 1.5 eq) was added. Reaction mixture was carried out at room temperature overnight (full conversion on TLC). K2CO3 was filtered off from reaction mixture and reaction was poured into ice water. Wh...
PUM
| Property | Measurement | Unit |
|---|---|---|
| Fraction | aaaaa | aaaaa |
| Composition | aaaaa | aaaaa |
Abstract
Description
Claims
Application Information
 Login to View More
Login to View More