

Antibacterial drugs cefoxitin preparation process
A technology for cefoxitin and cephalosporanic acid, applied in the field of compound preparation, can solve problems such as high production cost and low yield, and achieve the effects of saving cost, improving reaction yield and solving the problem of excessively low reaction yield
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

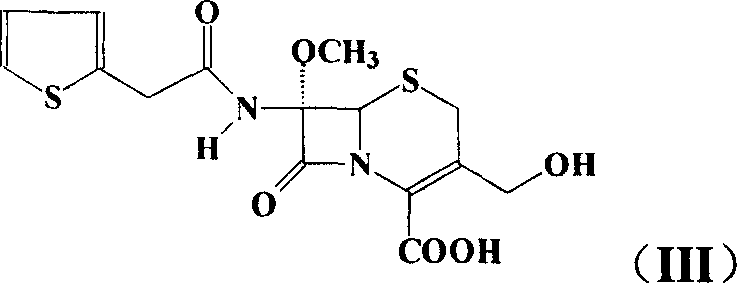
Examples
Embodiment 1
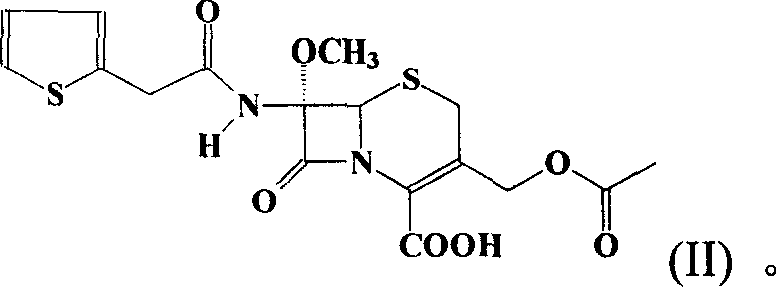
[0038] Step 1: Synthesis of 7α-methoxy-7β-[2-(2-thienyl)acetamido]-cephalosporanic acid (II)
[0039] Mix 30.2 g of 7α-methoxy-7β-amino-cephalosporanic acid (7-MAC) with 300 mL of water and cool to 0°C. Sodium bicarbonate was added portionwise with stirring until pH=7. A mixed solution of 24 g of thiopheneacetyl chloride and 30 mL of acetone was added dropwise within 1 hour, and saturated aqueous sodium carbonate solution was added dropwise at the same time to control the system to maintain pH=7-8. The reaction temperature is controlled between -5°C and 5°C. The reaction was monitored by high performance liquid chromatography until the starting material was consumed completely. After the reaction was completed, 900 mL of dichloromethane was added, and the pH was adjusted to 1-2 with 5% hydrochloric acid. Warm up to room temperature and separate the phases. The aqueous phase was extracted with appropriate amount of dichloromethane. The dichloromethane phases were combined ...
Embodiment 2
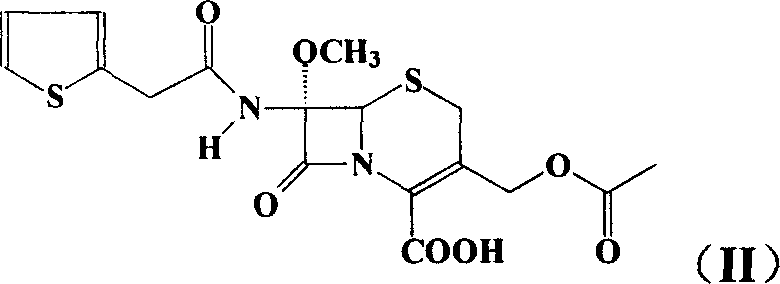
[0041] The first step: Synthesis of triethylamine salt of 7α-methoxy-7β-[2-(2-thienyl)acetamido]-cephalosporanic acid (II) 7α-methoxy-7β-amino-cephem Mix 30.2g of acid (7-MAC) with 300mL of water and cool to 0°C. Sodium bicarbonate was added portionwise with stirring until pH=7. A mixed solution of 24 g of thiopheneacetyl chloride and 30 mL of acetone was added dropwise within 1 hour, and saturated aqueous sodium carbonate solution was added dropwise at the same time to control the system to maintain pH=7-8. The reaction temperature is controlled between -5°C and 5°C. The reaction was monitored by high performance liquid chromatography until the starting material was consumed completely. After the reaction was completed, 900 mL of dichloromethane was added, and the pH was adjusted to 1-2 with 5% hydrochloric acid. Warm up to room temperature and separate the phases. The aqueous phase was extracted with appropriate amount of dichloromethane. The dichloromethane phases were...
Embodiment 3
[0043] Synthesis of Dibenzylamine Salt of 7α-Methoxy-7β-[2-(2-thienyl)acetamido]-cephalosporanic acid (II)
[0044] Mix 30.2 g of 7α-methoxy-7β-amino-cephalosporanic acid (7-MAC) with 300 mL of water and cool to 0°C. Sodium bicarbonate was added portionwise with stirring until pH=7. A mixed solution of 24 g of thiopheneacetyl chloride and 30 mL of acetone was added dropwise within 1 hour, and saturated aqueous sodium carbonate solution was added dropwise at the same time to control the system to maintain pH=7-8. The reaction temperature is controlled between -5°C and 5°C. The reaction was monitored by high performance liquid chromatography until the starting material was consumed completely. After the reaction was completed, 900 mL of dichloromethane was added, and the pH was adjusted to 1-2 with 5% hydrochloric acid. Warm up to room temperature and separate the phases. The aqueous phase was extracted with appropriate amount of dichloromethane. The dichloromethane phases ...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More