

Preparation method of tamsulosin
A technology for tamsulosin and compound, applied in the field of preparation of tamsulosin, can solve the problems of high price, difficult to obtain, difficult to purify, etc., and achieves the effects of low cost, high purity, and easy availability of raw materials
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

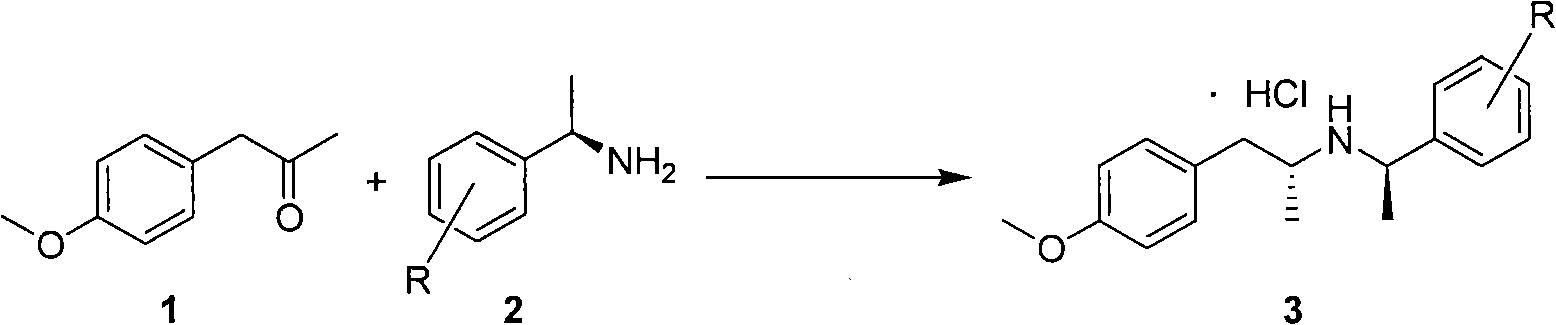
Examples
Embodiment 1
[0045] N-[(R)-(1-phenethyl)]-(R)-[1-(4-methoxyphenyl)propyl]-2-amine hydrochloride.
[0046] in N 2 Under protection, 250 mL of dichloromethane and 11.3 g of sodium borohydride were added to a 500 mL three-necked flask, and a mixture of 52.4 mL of glacial acetic acid and 40 mL of dichloromethane was slowly added dropwise at a temperature lower than 30°C. After dropping, react at room temperature for 1 hour until no gas emerges. This method prepares sodium triacetoxy borohydride (NaBH(OAc) 3 ). Dissolve 32.8g of compound 1 and 25.4g of compound 2 in 40mL of dichloromethane, and slowly add the above mixed solution to NaBH(OAc) 3 , and reacted at room temperature for 3 hours until compound 1 disappeared. After the reaction, the reaction solution was quenched with sodium hydroxide solution, and after liquid separation, the organic phase was dried and concentrated to obtain a colorless liquid. The liquid was dissolved in ethyl acetate, and hydrochloric acid ethanol solution was...
Embodiment 2
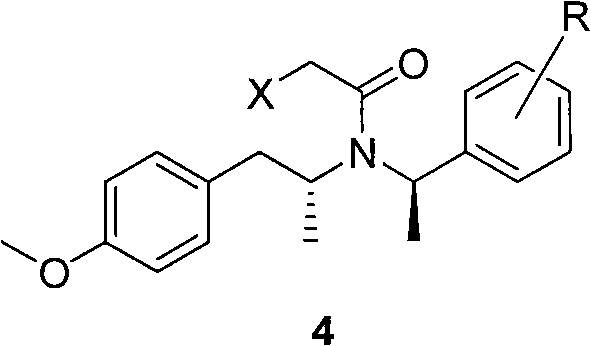
[0049] N-(R)-(1-phenethyl)-N-(R)-2-[1-(4-methoxy)phenyl]propyl-2-chloroacetamide.
[0050] Add 300mL of dichloromethane, 30.6g of compound 3 and 60g of sodium hydroxide into a 500mL three-neck flask, and slowly add 31.8mL of chloroacetyl chloride dropwise at a temperature lower than 25°C for reaction. After the dropwise addition, the reaction was continued for 0.5 hour until compound 3 was complete. The reaction solution was neutralized with hydrochloric acid, then separated, dried and concentrated, with a purity of 99.03%.
[0051] A small amount of the solution was concentrated to obtain a pale yellow oil.
[0052] 1 H-NMR (400mHz, CDCl 3 ): δ7.42(m, 5H); 6.63(d, 2H, J=8.0Hz); 6.38(d, 2H, J=7.6Hz); 5.15(bm, 1H); 4.20(s, 2H); 3.72 (s, 3H); 3.17-3.07 (bm, 2H); 2.34 (m, 1H); 1.66 (d, 3H, J=6.8Hz); 1.28 (d, 3H, J=6.4Hz).
Embodiment 3
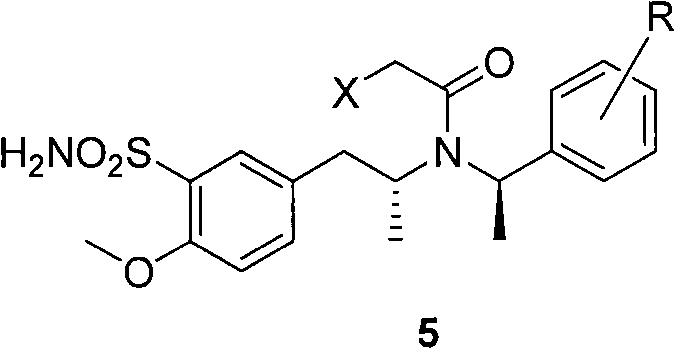
[0054] N-(R)-(1-phenethyl)-N-(R)-2-[1-(3-sulfonamido-4-methoxy)phenyl]propyl-2-chloroacetamide.
[0055] Add 66.0 mL of chlorosulfonic acid to a 250 mL three-necked flask with an acid scrubber, and slowly add 80.0 mL of compound 4 in dichloromethane dropwise at a temperature lower than 20°C, then react at room temperature for 5 hours until compound 4 disappeared completely. After the reaction was completed, the reaction solution was slowly poured into ice water to quench excess chlorosulfonic acid, then extracted twice with ethyl acetate, and the organic phase was concentrated to obtain a light yellow liquid. The liquid was dissolved in tetrahydrofuran, and 28.6 mL of 25-28% ammonia water was slowly added dropwise at a temperature lower than 25°C, and then reacted at room temperature for 1-2 hours until compound (5) disappeared. After the reaction was completed, tetrahydrofuran was removed by concentration, then water and ethyl acetate were added and fully dissolved by heatin...
PUM
| Property | Measurement | Unit |
|---|---|---|
| melting point | aaaaa | aaaaa |
Abstract
Description
Claims
Application Information
 Login to View More
Login to View More