

Collagen-integrin alpha2beta1 interacted polypeptide inhibitors and screening method thereof
A peptide inhibitor, collagen technology, applied in the direction of peptides, etc., can solve the problems of limited accuracy, time-consuming cost, fast calculation speed, etc.
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
Embodiment 1
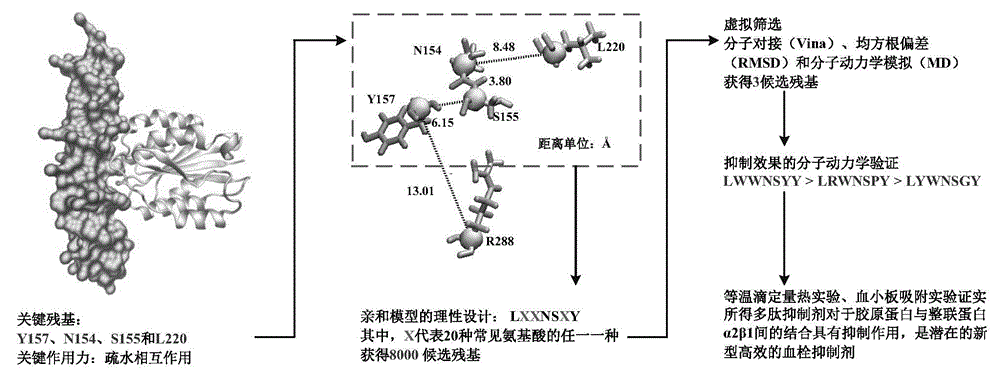
[0035] Example 1: Analysis of Collagen-Integrin α2β1 Interaction and Establishment of Peptide Inhibitor Library
[0036] First, the binding free energy of the α2A domain complex (PDB ID: 1DZI) in collagen-integrin α2β1 was calculated using the MM-PBSA method. Then, a free energy decomposition method based on MM-PBSA was used to analyze the molecular mechanism of the high affinity between collagen and α2A domain and to analyze the contribution of each residue in the collagen-α2A domain complex to the binding free energy. This identifies the key residues for their interactions. The criterion of <-2.5 kcal / mol was used to identify the residues that contributed more to the free energy, and it was found that only four residues, Y157, N154, S155 and R288, met the above requirements in α2A. Residue L220 contributes -1.9 kcal / mol to the free energy of the complex, which is slightly larger than -2.5 kcal / mol, but the contribution to the binding process follows Y157 and N154 so was als...
Embodiment 2
[0037] Embodiment 2: docking of polypeptide inhibitors and collagen fragments
[0038] Because a single residue has a strong affinity with collagen fragments, it does not necessarily mean that the polypeptide inhibitor composed of residues has a strong interaction with collagen fragments, so continue to use Vina to combine all the peptides in the peptide inhibitor library Inhibitors are sequentially docked with collagen fragments. Docking results showed that all peptide inhibitors could bind to collagen fragments, and the predicted binding free energy of all peptide inhibitors was between -7.2 and -3.7 kcal / mol. A total of 177 peptide inhibitors with binding free energy lower than -6.5 kcal / mol were selected.
[0039] In order to give full play to the affinity between key residues and collagen, it is necessary to search for peptide inhibitors whose docking conformation of key residues is consistent with that of the corresponding hot spot residues in α2A. Such peptides inhibit...
Embodiment 3
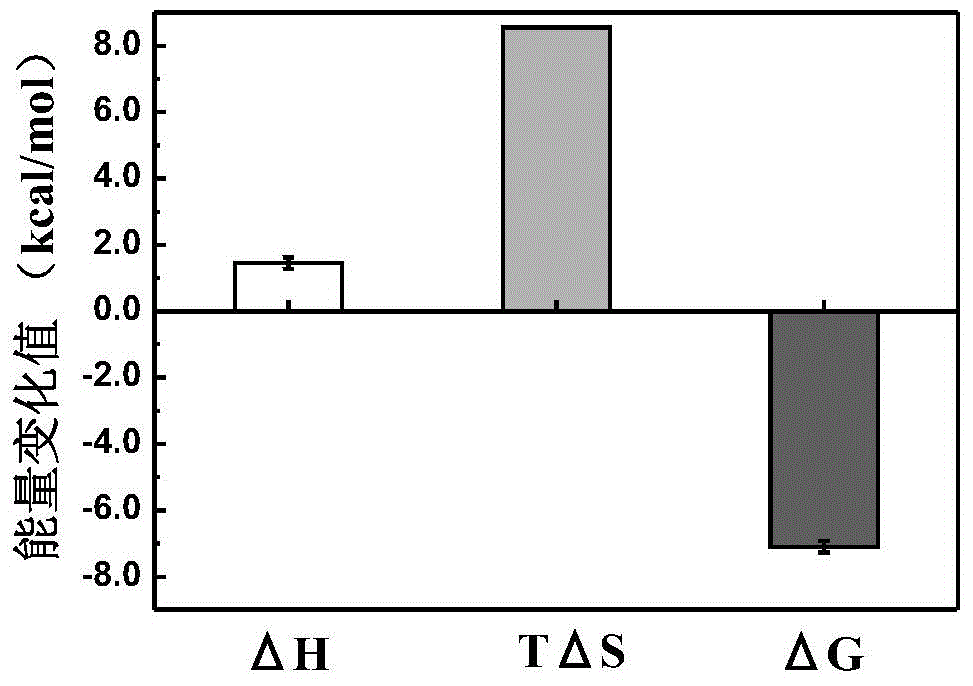
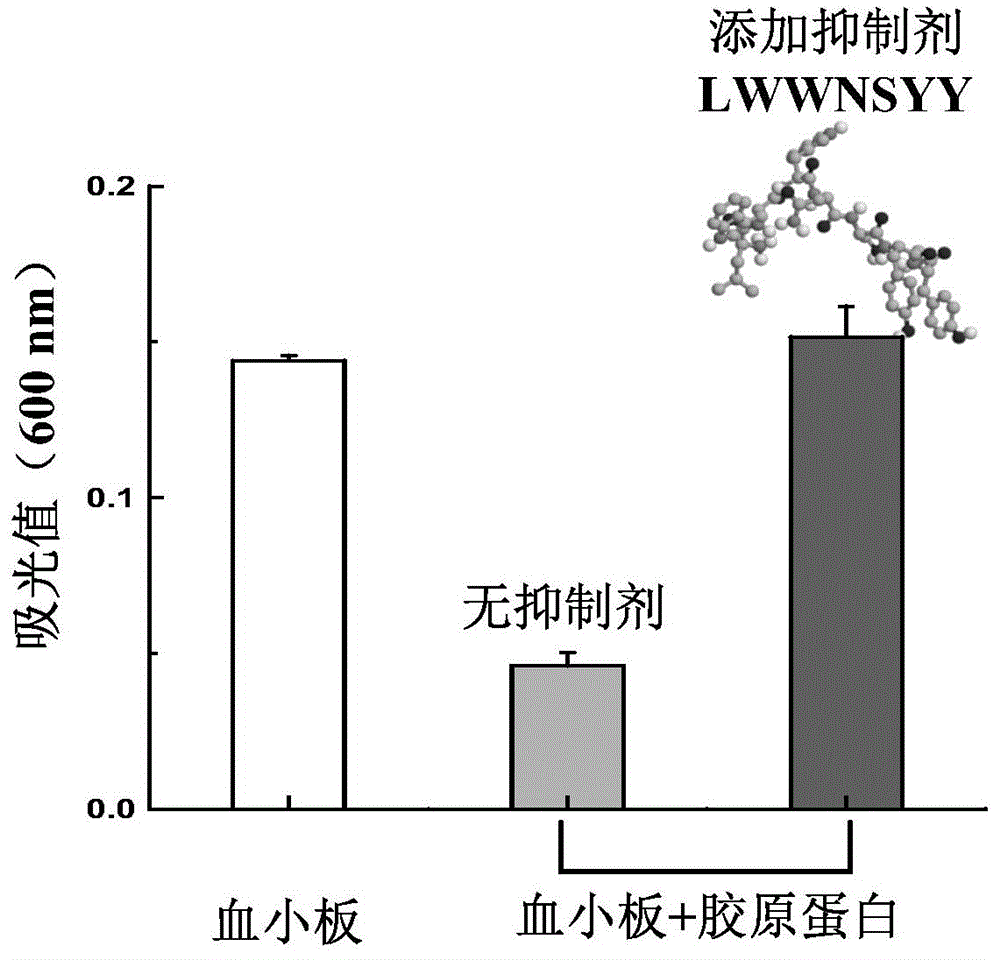
[0040] Example 3: Screening of polypeptide inhibitors and molecular dynamics simulation verification of their inhibitory effects
[0041] MD simulation is a very effective tool for studying protein dynamics. Protein fast internal motion, slow conformational change and folding process can all be studied using MD simulation. To further validate the collagen-peptide inhibitor interaction, not only its static structure (using molecular docking) but also its dynamic behavior should be studied. Therefore, MD simulation was used to study the dynamic information of the above-mentioned eight candidate peptide inhibitors binding to collagen fragments.
[0042] All MD simulations use the GROMACS 4.5.3 software package, select the G43a1 force field, and use the pdb2gmx command to convert the pdb coordinate structures of 8 candidate peptide inhibitors into the GROM structure dedicated to GROMACS; use the editconf command to convert the peptide inhibitors and collagen Placed at 10 x 7 x 5....
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More