

Process for producing pidotimod
A production process, the technology of pidotimod, applied in the direction of peptides, etc., can solve the problems of irritating eyes, skin and respiratory tract, low product yield and purity, and prominent environmental problems, so as to simplify the production process, improve the yield, Reduce the effect of intermediate links
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

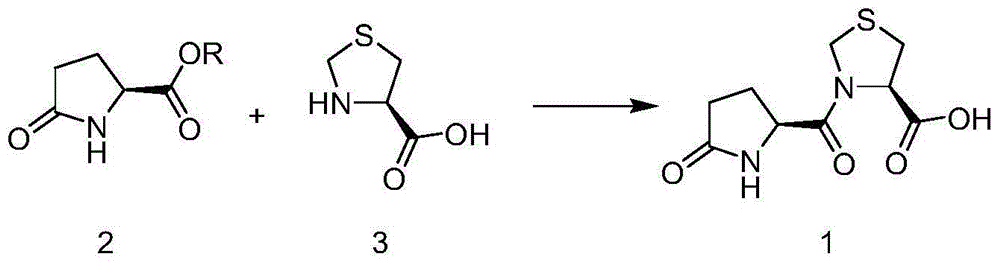
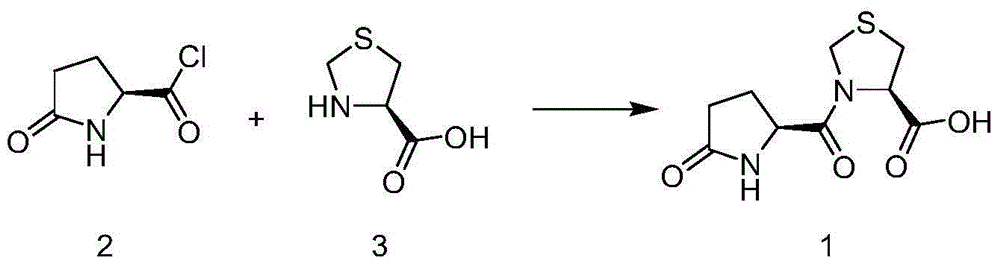
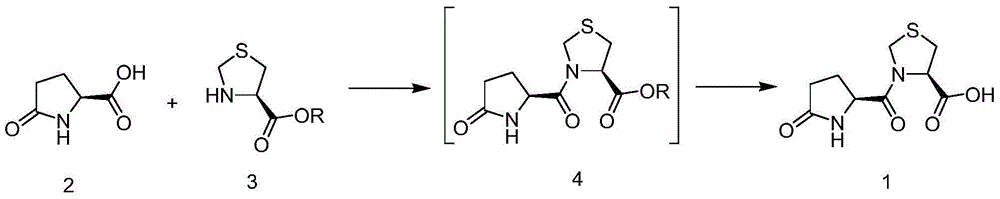
Examples
Embodiment 1
[0026] Under ice bath conditions, L-pyroglutamic acid (1.29g, 0.01mol), DCC (2.47g, 0.012mol) and methyl L-4-thiazolidinecarboxylate (1.33g, 0.01mol) were added to 25mL In ethyl acetate, after stirring for 0.5 hours, slowly warm up to room temperature, and continue to stir for 4 hours; add 1ml of water to terminate the reaction, stir for 15 minutes, filter the solid, and wash the solid with an appropriate amount of ethyl acetate, and the filtrate is not further treated with ethyl acetate spare.
[0027] Add 5ml of 1N dilute hydrochloric acid to the organic phase, keep stirring at room temperature for 2 hours, a large amount of solids precipitate out in the reaction solution, separate the liquids and keep the water phase; and transfer the water phase to an ice bath to continue stirring, and filter the precipitated solids in 4 hours to obtain White crystalline powder (1.46 g, yield: 59%, purity: 99.81%).
Embodiment 2
[0029] Under ice-bath conditions, L-pyroglutamic acid (1.29g, 0.01mol), DCC (3.09g, 0.015mol) and methyl L-4-thiazolidinecarboxylate (1.33g, 0.01mol) were added to 25mL In ethyl acetate, after stirring for 0.5 hours, slowly warm up to room temperature, and continue to stir for 4 hours; add 1ml of water to terminate the reaction, stir for 15 minutes, filter the solid, and wash the solid with an appropriate amount of ethyl acetate, and the filtrate is not further treated with ethyl acetate spare.
[0030] Add 5ml of 1N dilute hydrochloric acid to the organic phase, keep stirring at room temperature for 2 hours, a large amount of solids precipitate out in the reaction solution, separate the liquids and keep the water phase; and transfer the water phase to an ice bath to continue stirring, and filter the precipitated solids in 4 hours to obtain White crystalline powder (1.51 g, yield: 61%, purity: 99.83%).
Embodiment 3
[0032] Under ice-bath conditions, L-pyroglutamic acid (1.29g, 0.01mol), DCC (3.09g, 0.015mol), DMAP (0.006g) and methyl L-4-thiazolidinecarboxylate (1.33g, 0.01 mol) were added to 25 mL of ethyl acetate, and after stirring for 0.5 hours, the temperature was slowly raised to room temperature, and the stirring was continued for 2 hours; 1 ml of water was added to terminate the reaction, and after stirring for 15 minutes, the solid was filtered, and the solid was washed with an appropriate amount of ethyl acetate, and the filtrate acetic acid The ethyl ester was used without further treatment.
[0033] Add 5ml of 1N dilute hydrochloric acid to the organic phase, keep stirring at room temperature for 2 hours, a large amount of solids precipitate out in the reaction solution, separate the liquids and keep the water phase; and transfer the water phase to an ice bath to continue stirring, and filter the precipitated solids in 4 hours to obtain White crystalline powder (1.63 g, yield:...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More