

Methods for searching stable docking models of biopolymer-ligand molecule complex
a docking model and docking technology, applied in chemical methods analysis, instruments, molecular structures, etc., can solve the problems of inability to determine the binding modes of all ligand molecules of interest to their target biopolymer by experimental methods such as x-, inability to detect stable structures of complexes in experiments, and large analysis time and effort required for analysis. , to achieve the effect of greatly speeding up the docking model of biopolymer-ligand molecule complexes
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Benefits of technology
Problems solved by technology
Method used
Image

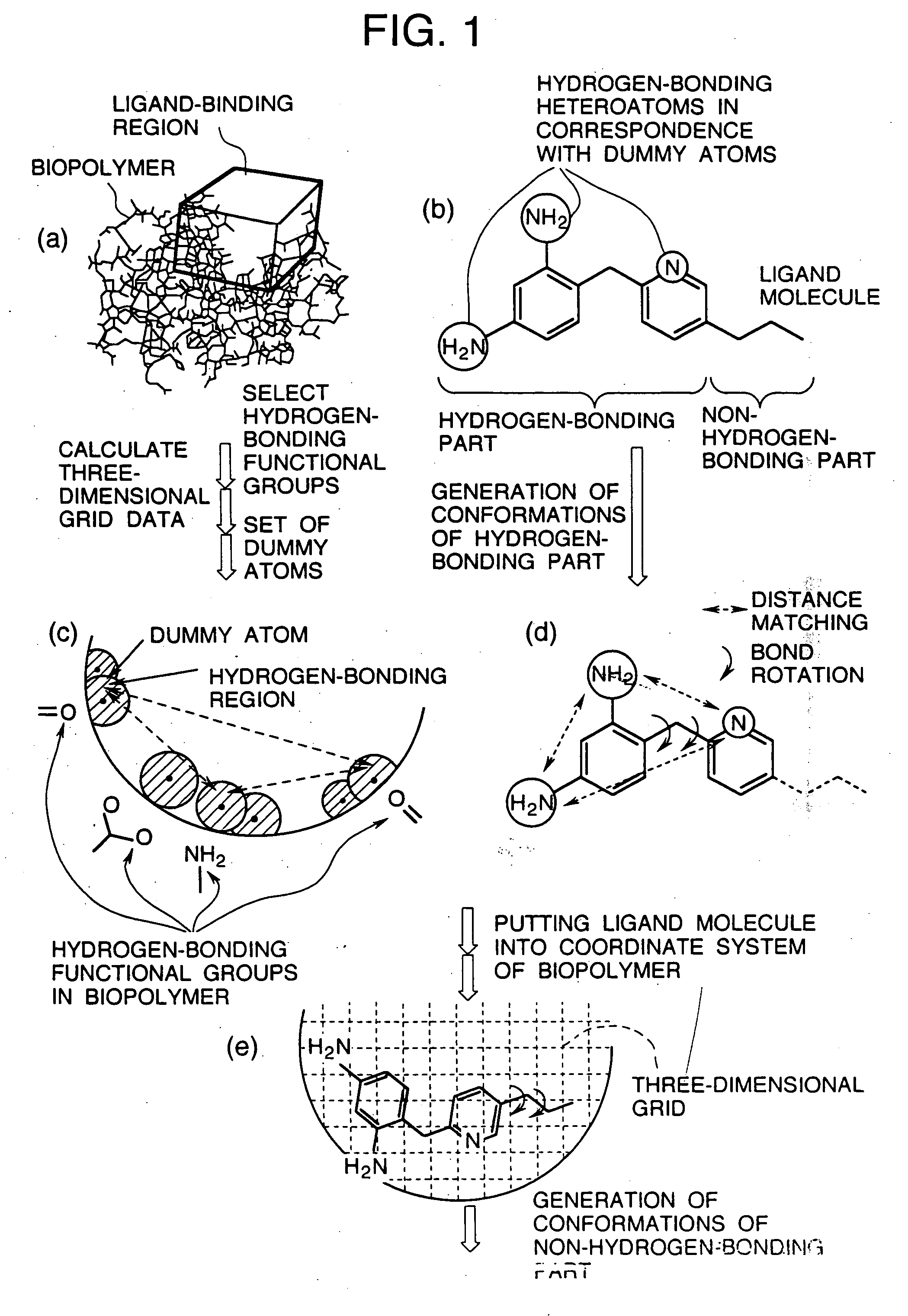
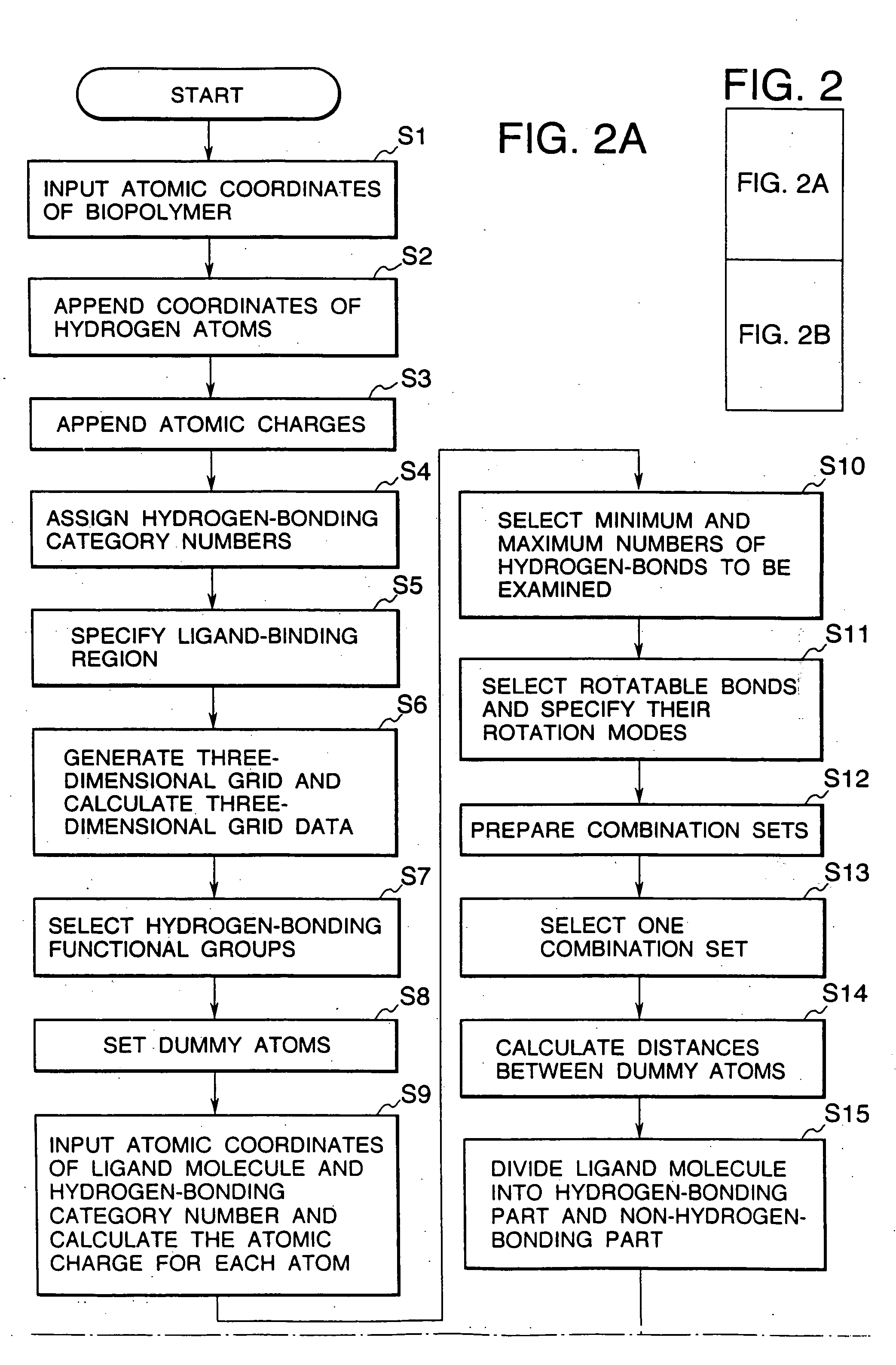
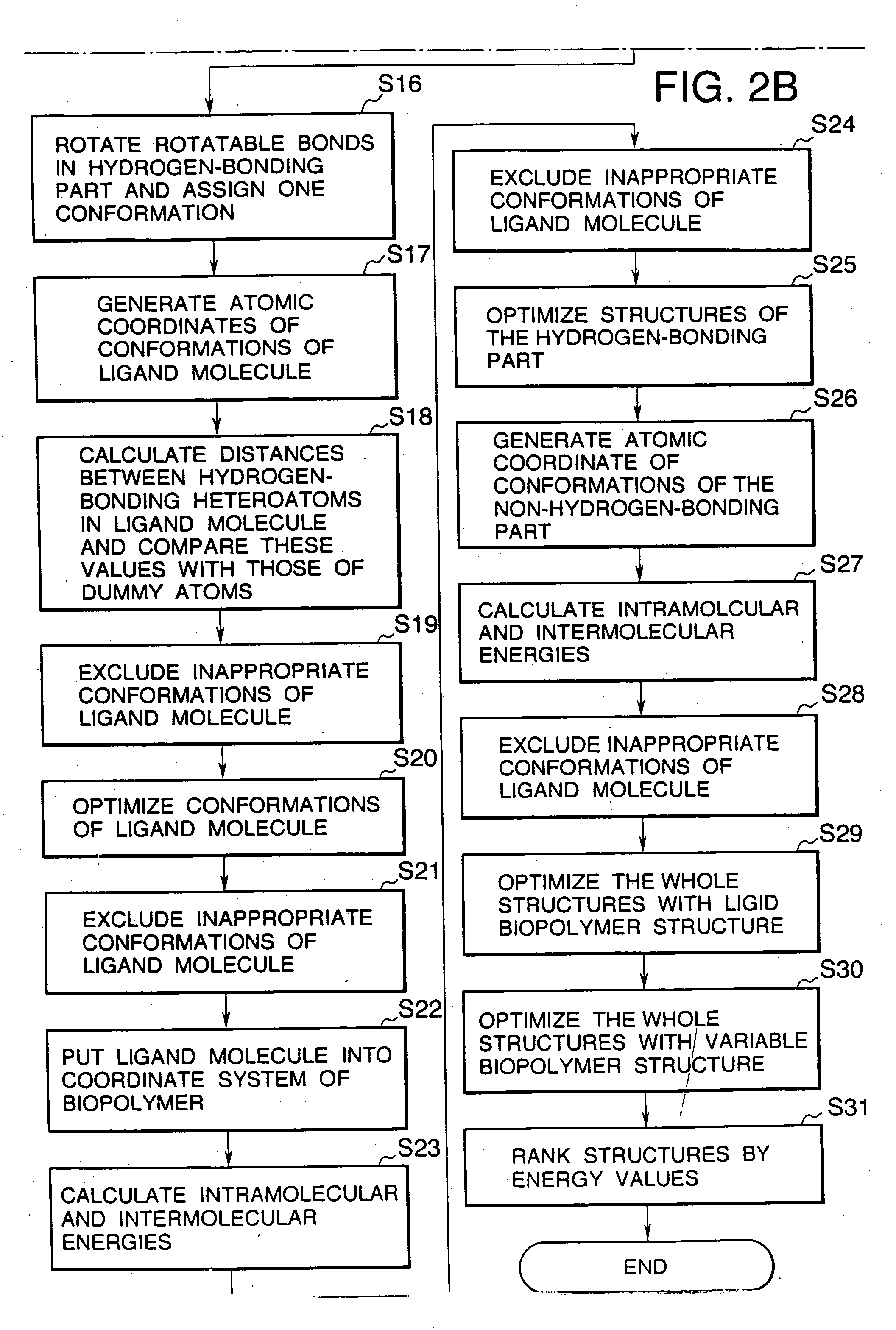
Examples
example 1
Methotrexate Molecule
[0128] The terminal carboxyl group of MTX molecule was removed to simplify the example and the following procedure was performed.
[0129] As the atomic coordinates of MTX, the atomic coordinates of the unbound crystal structure available from Cambridge Crystallographic Database were input. The atomic charge on each atom of MTX was calculated by the MNDO method in the MOPAC program. Since the nitrogen at position 1 in the pteridine ring of MTX was susceptible to protonation, the atomic charge thereon was calculated on the assumption that it was protonated. In the structures shown in FIG. 3, the encircled heteroatoms were selected as the hydrogen-bonding heteroatoms and the hydrogen-bonding category numbers were assigned thereto. In the MTX molecular structure shown in FIG. 3, bond a was rotated at intervals of 60° in the range of 0°-360°, bond b was rotated at intervals of 60° in the range of 0°-180°, bond c was assigned as either 0° or 180°, and bond d was set ...
example 2
Dihydrofolic Acid Molecule
[0137] Although the binding mode of DHFR to its substrate DHF has not yet been identified by X-ray analysis, it has been predicted from the stereospecifity of tetrahydrofolic acid that is the product of enzymatic reaction for the following reasons that the DHF molecule binds to the enzyme in a different mode from that of binding with MTX:
[0138] It is known that the hydrogen at position C6 in the tetrahydrofolic acid that is produced by the reducing action of DHFR is derived as a hydride ion from coenzyme NADPH. If it is assumed that the binding mode of the DHFR-DHF complex is the same as that of the MTX and DHFR molecules in the crystal structure of the ternary DHFR-MTX-NADPH complex obtained by X-ray analysis, tetrahydrofolic acid with opposite chirality should be produced. This strongly indicated that pteridine of DHF molecule is reversed from that of MTX (see FIG. 6).
[0139] In Example 2, the structures of stable DHFR-DHF complexes were searched witho...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More