

Method for binding site identification by molecular dynamics simulation (silcs: site identification by ligand competitive saturation)
a molecular dynamics and simulation technology, applied in the field of binding site identification by molecular dynamics simulation, can solve the problems of significant time, labor and material costs associated with these two biophysical fragment-based drug discovery approaches, and the computational approach is limited in its ability, so as to prevent non-polar aggregation and high probability, the effect of high binding probability
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Benefits of technology
Problems solved by technology
Method used
Image

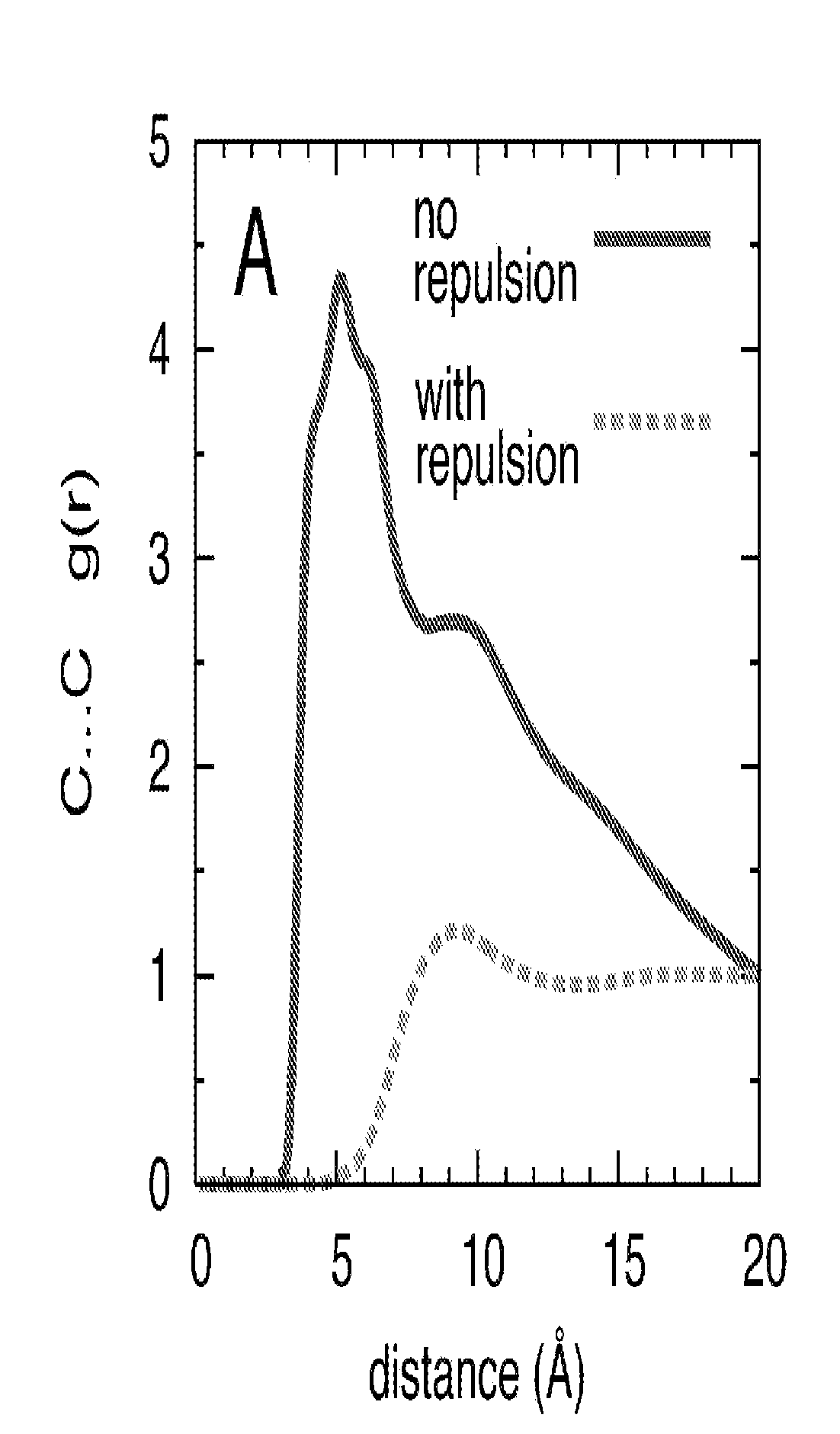
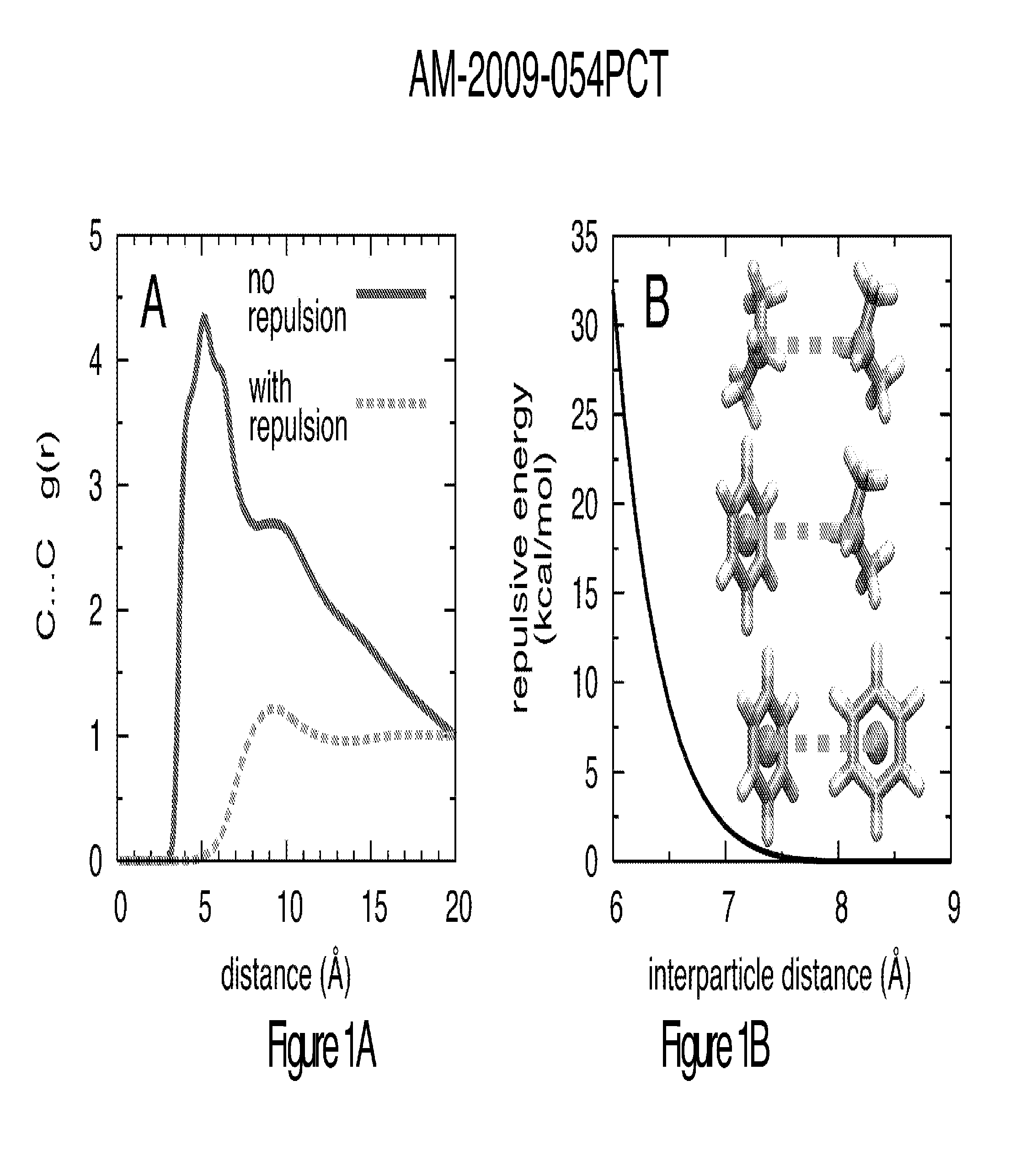

Examples
Embodiment Construction
[0046]The embodiments of the invention and the various features and advantageous details thereof are explained more fully with reference to the non-limiting embodiments of that are illustrated in the accompanying drawings and detailed in the following description. It should be noted that the features illustrated in the drawings are not necessarily drawn to scale. Descriptions of well-known materials, components, and processing techniques are omitted so as to not unnecessarily obscure the embodiments of the invention. The examples used herein are intended to merely facilitate an understanding of ways in which the embodiments of the invention may be practiced and to further enable those of skill in the art to practice the embodiments of the invention. Accordingly, the examples should not be construed as limiting the scope of the embodiments of the invention.
[0047]As stated above, there remains a need for a method of computational chemistry that identifies the binding site of fragments...
PUM
| Property | Measurement | Unit |
|---|---|---|
| concentration | aaaaa | aaaaa |
| 3D structure | aaaaa | aaaaa |
| repulsive interaction energy | aaaaa | aaaaa |
Abstract
Description
Claims
Application Information
 Login to View More
Login to View More