

Method for preparing sitafloxacin
A technology for sitafloxacin and compound, which is applied in the field of preparation of sitafloxacin, can solve the problems of many impurities, low yield, complicated post-processing and the like, and achieves the effects of high yield, reduced reaction rate and easy operation.
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

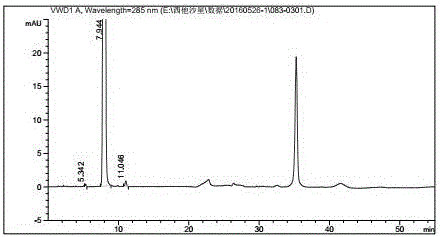
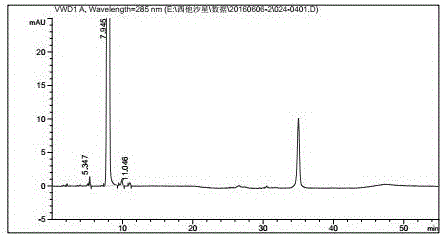
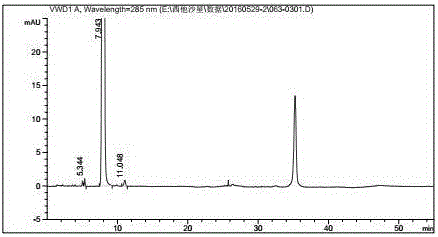
Examples
Embodiment 1
[0031] Dissolve 50.0 g of compound II and 40.0 g of compound III in 600 ml of acetonitrile, add 20.5 g of triethylamine, raise the temperature to 80° C., and react for 2 hours. TLC detects that the reaction is complete. Cool down to 0°C, stir for 2 hours, then filter with suction, and dry the filter cake with hot air at 45°C for 6 hours to obtain 70.2 g of pale yellow product IV. Yield: 87.4%.
[0032] Dissolve 50.0g of compound IV in 500ml of dichloromethane, and drop it into 250ml of 37% concentrated hydrochloric acid that has been cooled to 0°C in advance. During the dropwise addition, the temperature was controlled at 0°C to 5°C. After dropping, the temperature was raised to 20°C to 30°C for 1 hour. After the reaction is complete. Separate the water layer, adjust the pH value to 6∼7 with 20% sodium hydroxide, then adjust the pH value to 9∼10 with 28% concentrated ammonia water, concentrate under reduced pressure at room temperature to remove ammonia, and suction filter ...
Embodiment 2
[0035] Dissolve 100.0 g of compound II and 80.0 g of compound III in 1200 ml of acetonitrile, add 41.0 g of triethylamine, raise the temperature to 80° C., and react for 2 hours. TLC detects that the reaction is complete. Cool down to 0°C, stir for 2 hours, then filter with suction, and dry the filter cake with hot air at 45°C for 6 hours to obtain 143.0 g of pale yellow product IV. Yield: 89.0%.
[0036] 100.0g of compound IV was dissolved in 2000ml of ethyl acetate, and dropped into 1000ml of 37% concentrated hydrochloric acid which had been cooled to 0°C in advance. During the dropwise addition, the temperature was controlled at 0°C to 5°C. After dropping, the temperature was raised to 20°C to 30°C for 1 hour. After the reaction is complete. Separate the water layer, adjust the pH value to 2∼3 with 15% sodium hydroxide, then adjust the pH value to 9∼10 with 28% concentrated ammonia water, concentrate under reduced pressure at room temperature to remove ammonia, and sucti...
Embodiment 3
[0039] Dissolve 100.0 g of II and 80.0 g of III in 2000 ml of acetonitrile, add 41.0 g of triethylamine, raise the temperature to 80°C, and react for 2 hours. TLC detects that the reaction is complete. Cool down to 0°C, stir for 2 hours, then filter with suction, and dry the filter cake with hot air at 45°C for 6 hours to obtain 147.0 g of pale yellow product IV. Yield: 91.5%.
[0040] 100.0 g of compound IV was dissolved in 1600 ml of methyl acetate, and dropped into 800 ml of 37% concentrated hydrochloric acid that had been cooled to 0°C in advance. During the dropwise addition, the temperature was controlled at 0°C to 5°C. After dropping, the temperature was raised to 20°C to 30°C for 1 hour. After the reaction is complete. Separate the water layer, adjust the pH value to 6∼7 with 10% potassium hydroxide, then adjust the pH value to 10∼11 with 28% concentrated ammonia water, concentrate under reduced pressure at room temperature to remove ammonia gas, and suction filter ...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More