Cathepsin K inhibitors and application thereof
A technology of compounds and atoms, applied in the direction of anti-inflammatory agents, drug combinations, non-central analgesics, etc., can solve the problems of unsatisfactory selective inhibition of various cathepsins and disturbing side effects
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

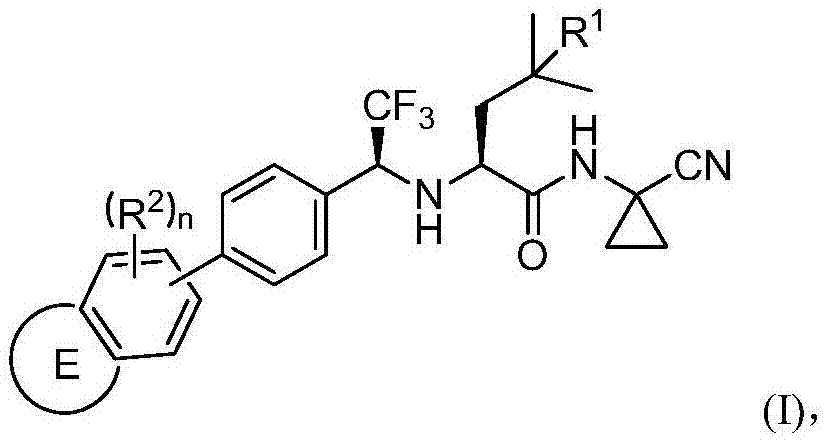
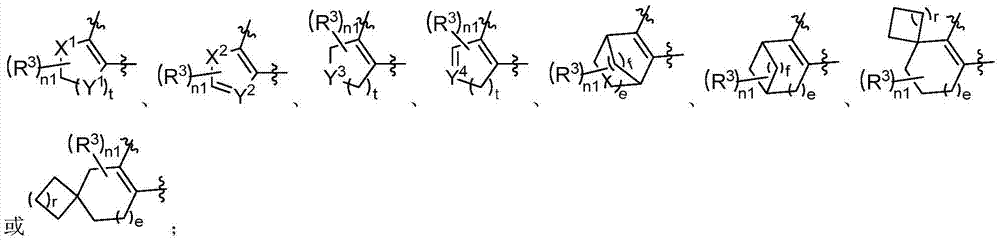
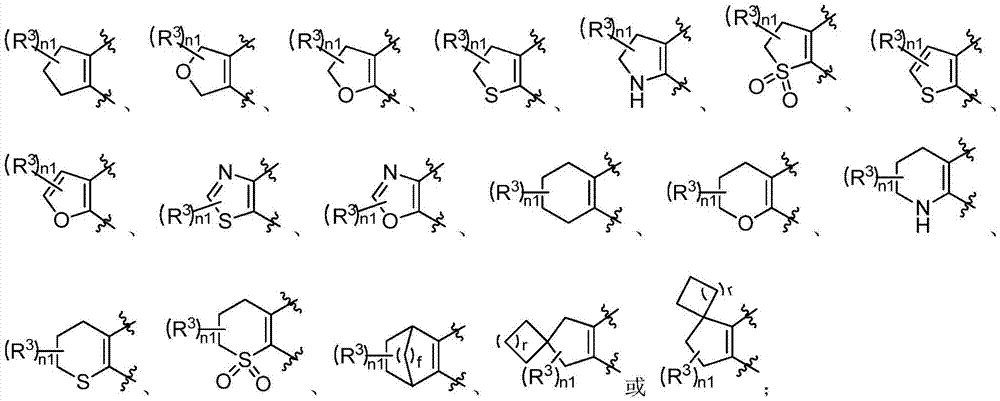
Examples
Embodiment 1
[0191] (S)-N-(1-cyanocyclopropyl)-4-fluoro-4-methyl-2-(((S)-2,2,2-trifluoro-1-(4-(4' -(2-oxopyrrolidin-1-yl)-1',3'-dihydrospiro[cyclopentane-1,2'-indene]-7'-yl)phenyl)ethyl)amino)pentyl Amide
[0192]
[0193] Step 1: 4'-(2-oxopyrrolidin-1-yl)-1',3'-dihydrospiro[cyclopentane-1,2'-indene]-7'-yltrifluoromethanesulfonic acid ester
[0194] Add 4'-iodo-1',3'-dihydrospiro[cyclopentane-1,2'-indene]-7'-yl triflate (522mg, 1.77 mmol) [refer to the synthesis method of patent WO 2014082379, the synthetic route of intermediate 1-8 on page 113-115], 2-pyrrolidone (151mg, 1.77mmol), potassium phosphate (0.75g, 3.54mmol), cuprous iodide ( 17mg, 0.09mmol) and anhydrous toluene (10mL), N,N'-dimethylethylenediamine (8.0mg, 0.09mmol) was added under nitrogen flow, sealed and heated to 130°C for overnight reaction. After the reaction was completed, it was cooled to room temperature, quenched with water, extracted with ethyl acetate (30 mL×2), the organic phase was washed with saturated br...
Embodiment 2
[0200] (2S)-N-(1-cyanocyclopropyl)-4-fluoro-4-methyl-2-(((1S)-2,2,2-trifluoro-1-(4-(8- (2-Oxopyrrolidin-1-yl)-1,2,3,4-tetrahydro-1,4-methyconaphthalene-5-yl)phenyl)ethyl)amino)pentanamide
[0201]
[0202] Step 1: 5-Methoxy-1,4-dihydro-1,4-methaphthalene
[0203] Add magnesium chips (600mg, 24.9mmol), a small particle of elemental iodine and anhydrous tetrahydrofuran (10mL) to the dry reaction flask, install a reflux condenser, and protect it with nitrogen, first add 2-bromo-1-fluoro-3 -Methoxybenzene (0.5g) after initiation of the reaction, additional 2-bromo-1-fluoro-3-methoxybenzene (2.5g, 12.2mmol) and freshly distilled 1,3-cyclopentadiene (1.05g.17.56mmol) was dissolved in anhydrous tetrahydrofuran (20mL) and added slowly in a constant pressure dropping funnel to keep the reaction system slightly refluxed. After the dropwise addition was completed, the reaction system continued to be heated under reflux for 1 hour. After cooling, the reaction was quenched with satur...
Embodiment 3
[0225] (S)-N-(1-cyanocyclopropyl)-4-fluoro-4-methyl-2-(((S)-2,2,2-trifluoro-1-(4-(4' -(2-oxopyrrolidin-1-yl)-2',3'-dihydrospiro[cyclopropane-1,1'-indene]-7-yl)phenyl)ethyl)amino)pentanamide
[0226]
[0227] Step 1: 7-Methoxy-1-methylene-2,3-dihydro-1H-indene
[0228] Potassium tert-butoxide (3.50g, 30.86mmol), methyltriphenylphosphine iodide (13.7g, 33.9mmol) and anhydrous tetrahydrofuran (200mL) were added to the reaction flask, and protected with nitrogen, reacted at room temperature for 1 hour . Then a solution of 7-methoxy-2,3-dihydro-1H-inden-1-one (5.0g, 30.86mmol) in anhydrous tetrahydrofuran (20mL) was added dropwise to the reaction flask, and after the addition was complete, the reaction mixture Stir overnight at room temperature. After the reaction was complete, it was quenched by adding water (80 mL), extracted with ethyl acetate (80 mL×3), and the organic phase was washed with saturated brine (60 mL), and dried over anhydrous sodium sulfate. The solvent was...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More - Generate Ideas
- Intellectual Property
- Life Sciences
- Materials
- Tech Scout
- Unparalleled Data Quality
- Higher Quality Content
- 60% Fewer Hallucinations
Browse by: Latest US Patents, China's latest patents, Technical Efficacy Thesaurus, Application Domain, Technology Topic, Popular Technical Reports.
© 2025 PatSnap. All rights reserved.Legal|Privacy policy|Modern Slavery Act Transparency Statement|Sitemap|About US| Contact US: help@patsnap.com