

Analysis method and system of metagenome data
A metagenomic and data analysis technology, applied in the field of bioinformatics, can solve problems such as poor specificity, many false positives in test results, and shorten analysis time, and achieve the effect of reducing the amount of calculation, eliminating false positive results, and controlling the calculation time.
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
Embodiment 1
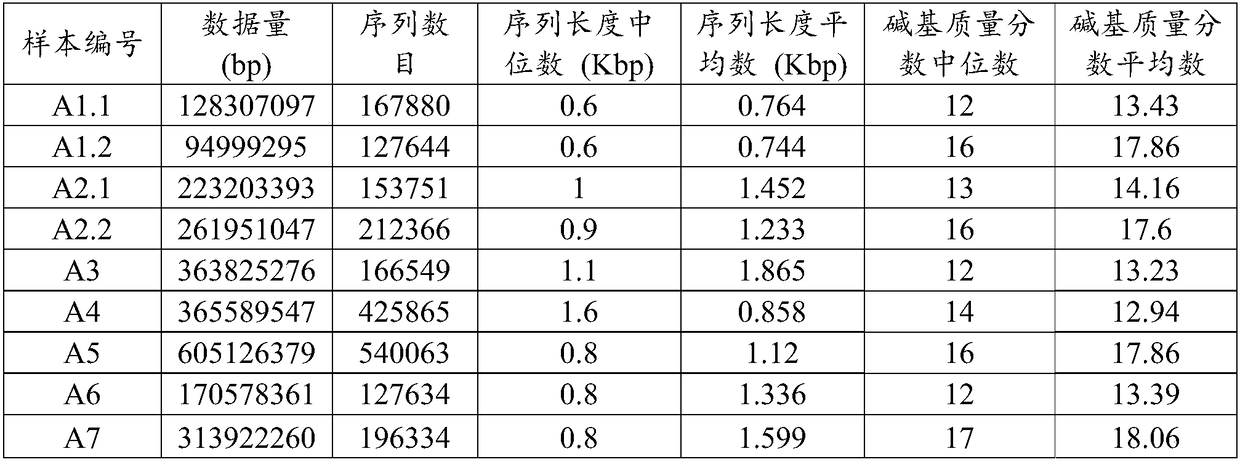
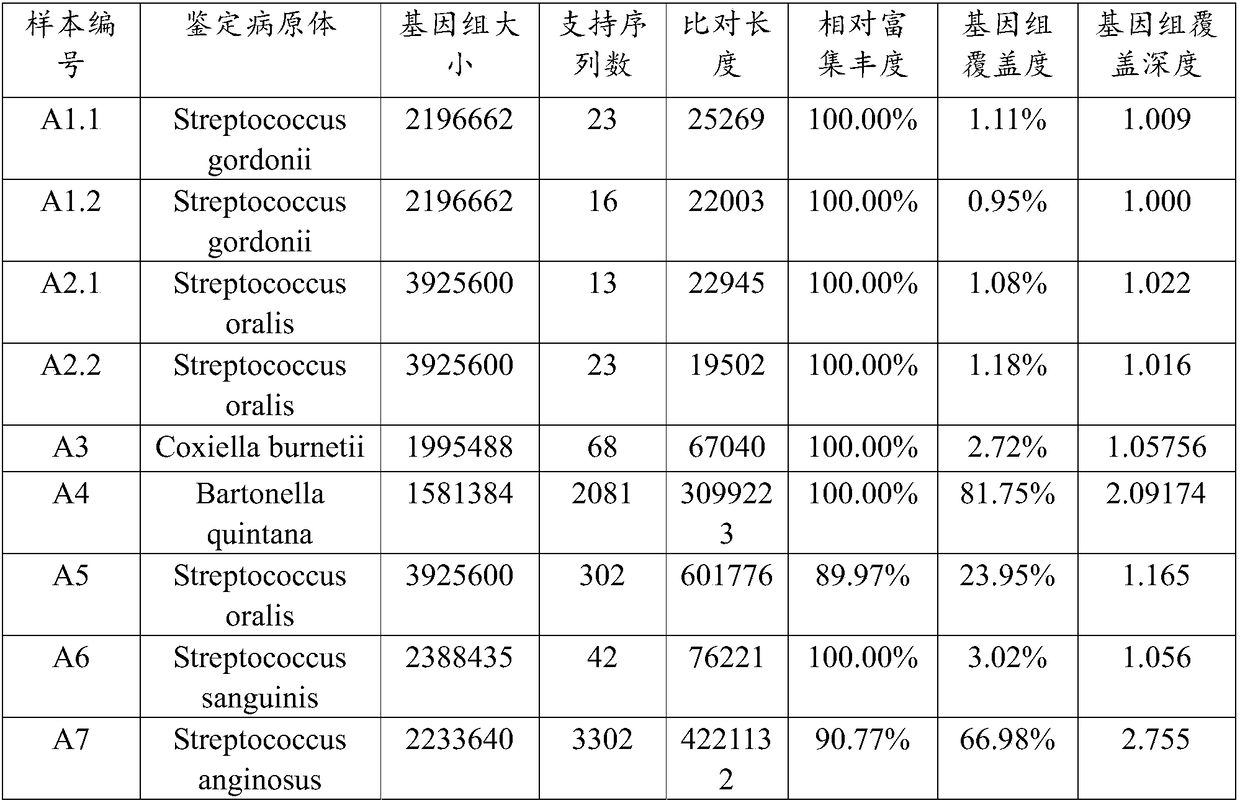
[0082] Example 1 Metagenome detection and data analysis of cardiac neoplasm samples based on Nanopore sequencing platform
[0083] The cardiac vegetation samples A1-A7 were collected from 7 cases of infective endocarditis patients with negative clinical culture for valve replacement surgery, and stored in a -80°C refrigerator.
[0084] Nucleic acid was extracted from the obtained samples according to the following procedure: take out the neoplastic sample from the refrigerator and place it at room temperature for 30 minutes, then use sterilized scissors to cut the neoplastic sample into pieces, and use the TIANamp Micro DNA kit to perform nucleic acid extraction according to the instructions.
[0085] The extracted nucleic acid samples were constructed and sequenced according to the following procedures. The library construction scheme selected the 1D Native barcoding protocol provided by Oxford Nanopore:
[0086] 1) Use g-TUBE (Covaris) to disrupt 1.2 μg nucleic acid sample at ...
Embodiment 2
[0107] Example 2 Metagenome detection and data analysis of cardiac neoplasm samples based on Ilumina sequencing platform
[0108] Using A1-A2 in Example 1 as samples, genomic nucleic acid was extracted and a library was constructed, and Illumina HiseqPE150 was used for sequencing. After removing adapters and sequences with a high N ratio, sequence information in fastq format was obtained from the obtained sequencing data. The data analysis of each sample was carried out as follows:
[0109] 1) The data in fastq format generated by Ilumina sequencing is removed from the adapters and sequences with a high N ratio, and then enters the next step of quality assessment analysis.
[0110] 2) Sequencing quality identification. The read length of this data is 150, and the sequences whose length is less than 100bp and whose average sequencing quality is less than 25 are filtered out. If the GC ratio of the first 10 bases of the data is abnormal, the first 10 bases of each sequence will...
Embodiment 3
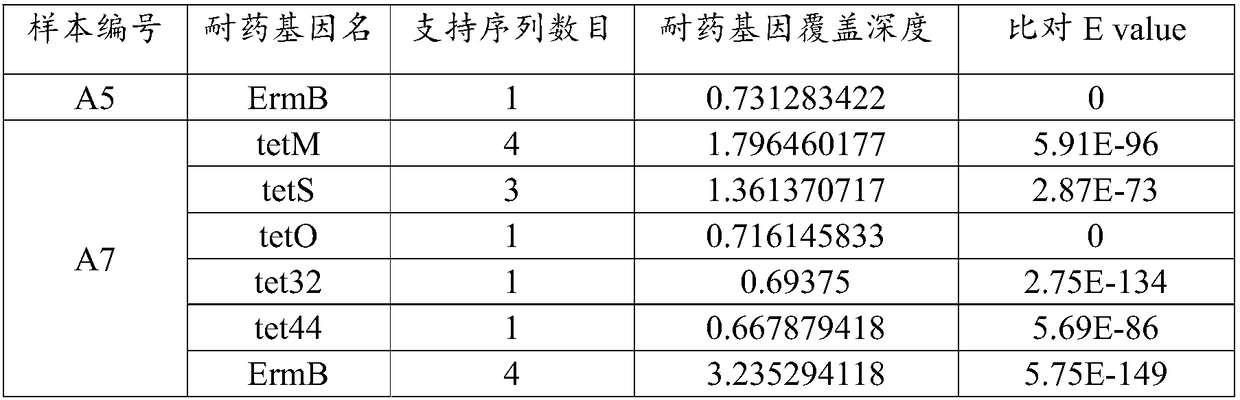
[0123] Example 3 Drug-resistant gene detection of postcardiac neoplasm samples based on BGI sequencing platform
[0124] Taking A1-A2 in Example 1 as samples, extract genomic nucleic acid and construct a library, use the BGI sequencing platform for sequencing, and perform the following data analysis on the data generated by BGI sequencing for each sample:
[0125] 1) The data in fastq format generated by BGI sequencing is removed from the adapters and sequences with a high N ratio, and then enters the next step of quality assessment analysis.
[0126] 2) Sequencing quality identification. The read length of the data library was 150, and the sequences with a length of <100bp and an average sequencing quality of <25 were filtered out.
[0127] 3) Remove the host sequence. By aligning to the human genome (genome version HG38), the sequences that failed to align were retained and entered into the next step of analysis.
[0128] 4) The "two-step method" is used to identify the p...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More