

Preparation method of Pranlukast intermediate
An intermediate and reaction time technology, applied in the field of pranlukast intermediates, can solve the problems of harsh reaction conditions and difficulty in industrial production, and achieve the effects of mild reaction conditions, low production cost and simple post-treatment
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

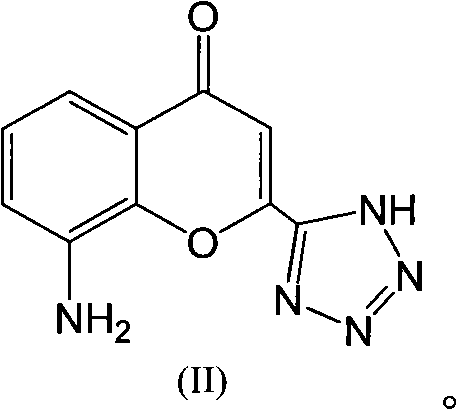
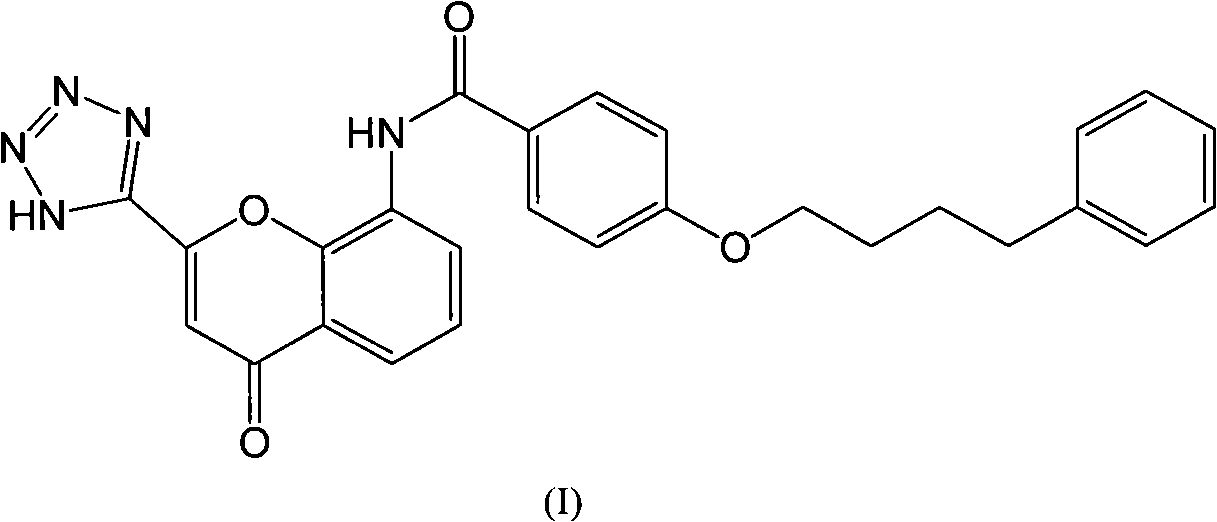
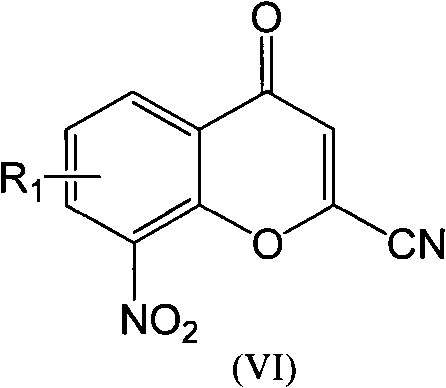
Examples
Embodiment 1
[0029] Example 1: Preparation of ethyl 2-formate-6-bromo-8-nitro-4-oxo-4H-1-benzofuran
[0030] Add 200ml of absolute ethanol to a 500ml flask, weigh 4.8g of sodium, and add sodium to the ethanol. After the metal sodium reacted completely, heat up to reflux, add 20ml of diethyl oxalate, 10g (38.6mmol) of 5-bromo-2-hydroxy-3-nitroacetophenone, heat up to 75°C and reflux. After 40 minutes, pour the reaction system into 240ml of 2N hydrochloric acid and stir for one hour. A large amount of khaki flocculent precipitates are produced. After filtering, add the solid to 120ml of ethanol system, add about 6ml of concentrated hydrochloric acid, heat up to reflux, and stir for reaction After about 60 minutes, it was cooled down to room temperature and filtered to obtain 10.35 g (30.3 mmol) of the target product. Yield 78.6%, Mp: 140-142°C. MS(m / z): 342.1(M+1), 364.1(M+23); 1 HNMR (CDCl 3 -d 1): δ8.58 (d, 1H), 8.48 (d, 1H), 7.21 (s, 1H), 4.47-4.51 (m, 2H), 1.44-1.47 (t, 3H).
Embodiment 2
[0031] Example 2: Preparation of 2-formamide-6-bromo-8-nitro-4-oxo-4H-benzofuran
[0032] Add the 6-bromo-8-nitro-4-oxo-4H-1-benzofuran-2-carboxylic acid ethyl ester obtained in the previous step reaction into 225ml of methanol, and pass through the ammonia gas by bubbling at room temperature , after reacting for 1.5h, the system was evaporated to dryness under reduced pressure to obtain a yellow solid, which was added to 150ml of concentrated hydrochloric acid, heated to 30-40°C and stirred for 2h, then diluted with 1000ml of ice water, filtered and the obtained filter cake was used Add it into 10% aqueous sodium bicarbonate solution and stir for 4-5 hours, filter and dry to obtain 6.44 g (0.021 mol) of the product, with a yield of 68.0%. Mp: 255-258°C; MS (m / z): 312.7 (M+1); 1 HNMR (DMSO-d 6 ): δ8.75(d, 1H), 8.42(d, 1H), 8.30(s, 1H), 8.00(s, 1H), 6.99(s, 1H).
Embodiment 3
[0033] Example 3: Preparation of 2-cyano-6-bromo-8-nitro-4-oxo-4H-1-benzopyran
[0034] Add POCl dropwise to 115ml DMF 3 9ml, the temperature is controlled at about 0°C, after the dropwise addition, stir and react at room temperature for 30min, dissolve the amide compound obtained above with 30mlDMF, add it to the reaction system, react at room temperature for 3h, and then slowly add the reaction system dropwise to 500ml ice In the water, solids were precipitated, filtered, the obtained product was added to 100ml ethyl acetate solution to dissolve the crude product, and 10% aqueous sodium bicarbonate solution was added dropwise until no solids were precipitated, filtered, the filtrate was allowed to stand for stratification, and organic The layer was washed with saturated brine and dried over anhydrous magnesium sulfate. After filtration, 0.5 g of activated carbon was added to the filtrate, and heated to reflux for half an hour. After removing the activated carbon, the filtrat...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More