Plants having increased yield-related traits and a method for making the same
A plant, yield technology, applied in the field of molecular biology, can solve the problems of average yield decline, crop loss, etc.
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image


Examples
Embodiment 1
[0519] Example 1: Identification of sequences related to nucleic acid sequences used in the methods of the invention
[0520] Using database sequence search tools such as the Basic Local Alignment Tool (BLAST) (Altschul et al. (1990) J. Mol. Biol. 215:403-410; and Altschul et al. (1997) Nucleic Acids Res. 25:3389-3402), Sequences (full-length cDNA, EST or genomic sequences) related to the nucleic acid sequences used in the methods of the invention were identified in the sequences maintained by the National Center for Biotechnology Information (NCBI) Entrez nucleotide database. The BLAST program is used to find regions of local similarity between sequences by comparing nucleic acid sequences or polypeptide sequences to sequence databases and by calculating the statistical significance of the matches. For example, the TBLASTN algorithm is applied to the polypeptide encoded by the nucleic acid sequence of the present invention, with the default setting and the filter turned on to...
Embodiment 2
[0526] Embodiment 2: Alignment of polypeptide sequences of the present invention
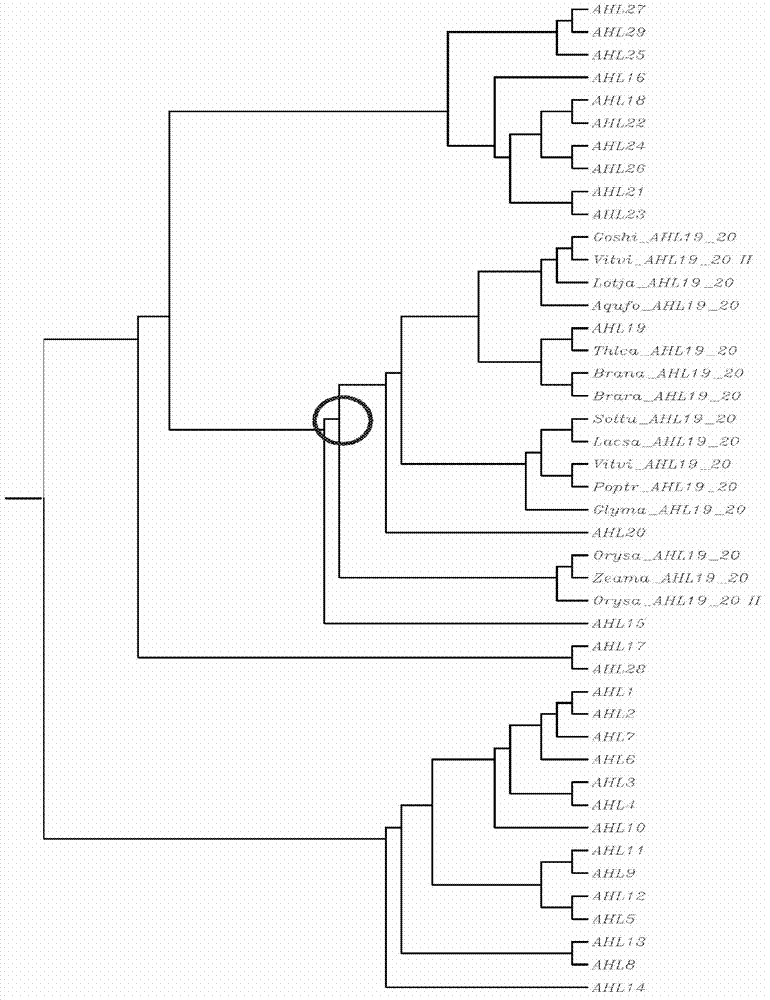
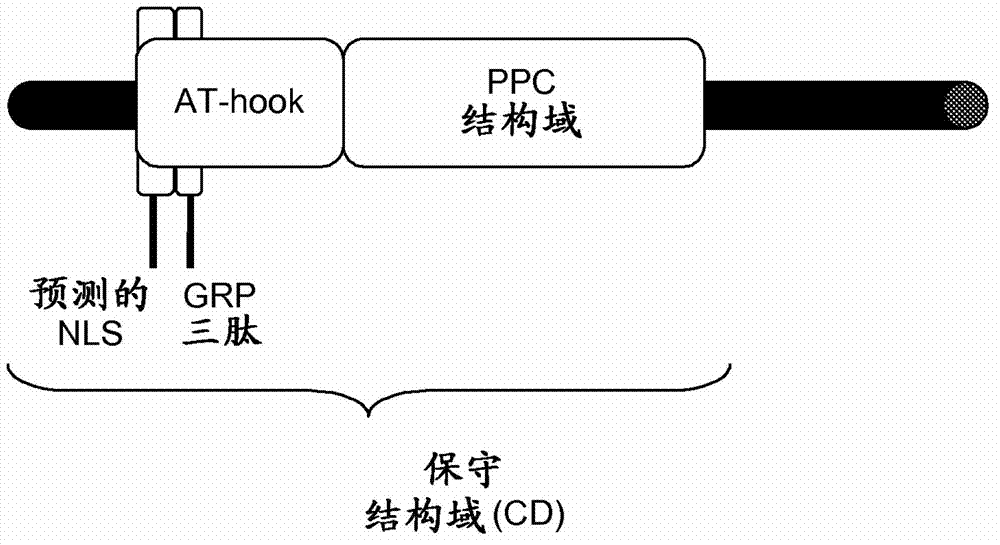
[0527] Polypeptide alignment was performed using the Vector NTI package (Invitrogen) AlignX program based on the popular progressive alignment ClustalW algorithm (Thompson et al (1997) Nucleic Acids Res 25:4876-4882; Chenna et al (2003) Nucleic Acids Res 31:3497-3500). Alignment of sequences. The default values are gap opening penalty of 10, gap extension penalty of 0.1, and the selected weight matrix is Blosum62 (if comparing peptides). Minor artificial edits can be made to further refine the alignment. A phylogenetic tree of polypeptides was constructed using the neighbor-joining clustering algorithm provided in the AlignX program of Vector NTI (Invitrogen).
[0528] Use progressive comparison Clustal algorithm (1.83), utilize default value (Thompson et al., (1997) Nucleic Acids Res25:4876-4882; Chenna et al., (2003).Nucleic Acids Res31:3497-3500), to Fujimoto etc. (2004 ; All Arabidops...
Embodiment 3
[0535] Example 3: Calculation of Global Identity Percentage Between Polypeptide Sequences That Can Be Used to Implement the Methods of the Invention
[0536] The global similarity and identity percentages between the full-length polypeptide sequences that can be used to implement the method of the present invention utilize one of the methods available in the art, i.e. MatGAT (Matrix Global Alignment Tool) software (BMC Bioinformatics.20034:29.MatGAT: An application that generates similarity / identity matrices using protein or DNA sequences. Campanella JJ, Bitincka L, Smalley J; software hosted by Ledion Bitincka), was identified. MatGAT software can generate similarity / identity matrices of DNA or protein sequences without pre-aligning the data. The program performs a series of pairwise alignments using the Myers and Miller global alignment algorithm (with a gap opening penalty of 12 and a gap extension penalty of 2) to calculate similarity and identity using, for example, Blosu...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More - R&D
- Intellectual Property
- Life Sciences
- Materials
- Tech Scout
- Unparalleled Data Quality
- Higher Quality Content
- 60% Fewer Hallucinations
Browse by: Latest US Patents, China's latest patents, Technical Efficacy Thesaurus, Application Domain, Technology Topic, Popular Technical Reports.
© 2025 PatSnap. All rights reserved.Legal|Privacy policy|Modern Slavery Act Transparency Statement|Sitemap|About US| Contact US: help@patsnap.com