

Synthesis method of metaraminol bitartrate
A technology of meta-hydroxylamine bitartrate and a synthesis method is applied in the synthesis field of meta-hydroxylamine bitartrate, can solve the problems of limited industrial application, high technical difficulty, many key technologies and the like, and achieves easy purchase, high enantioselectivity and high efficiency Effect
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

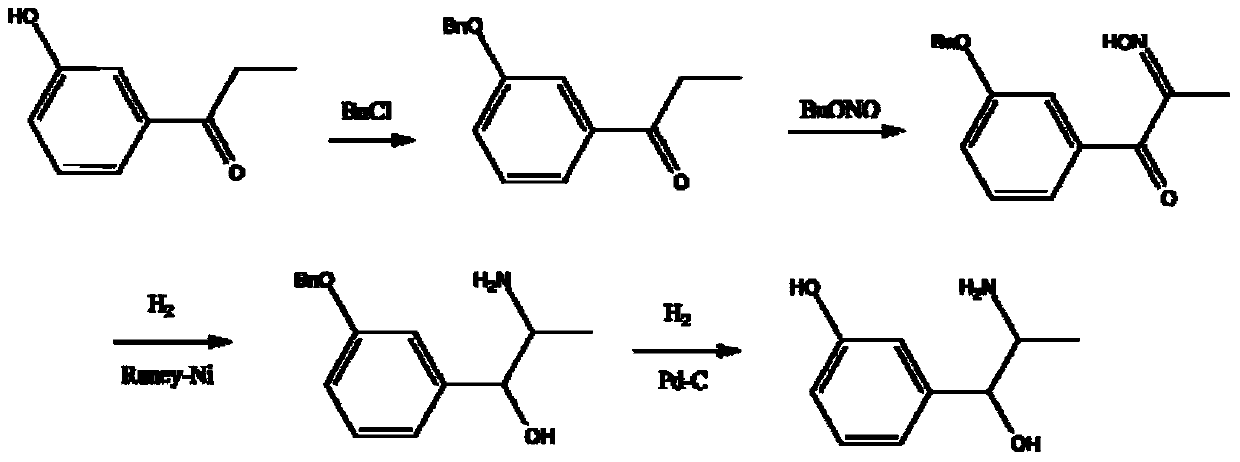
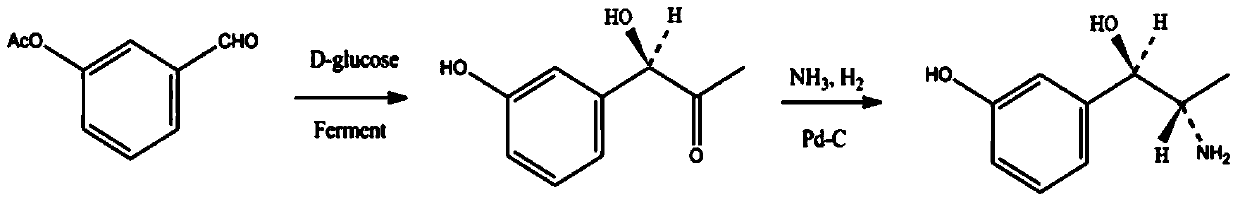
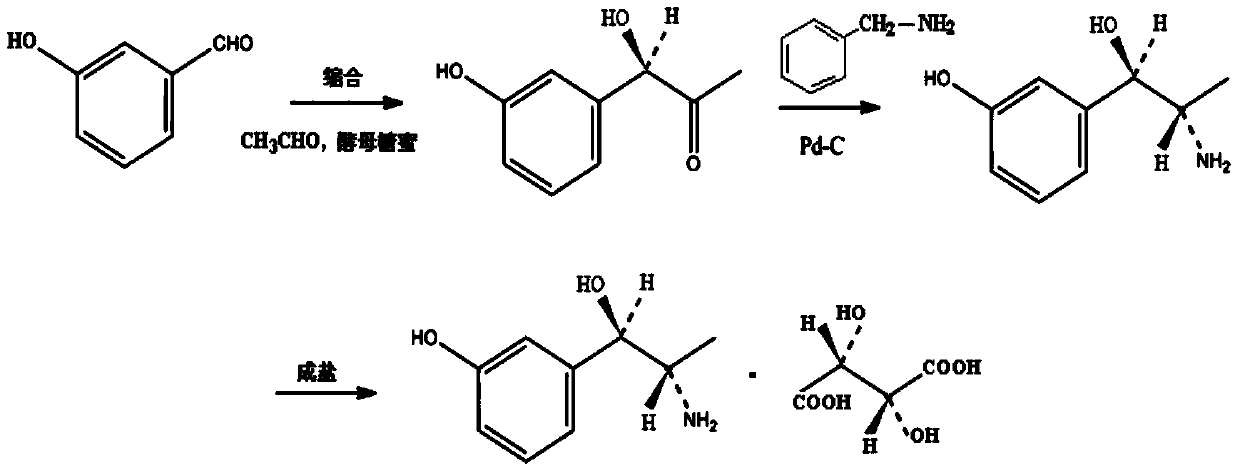
Examples
Embodiment 1
[0040] Weigh 0.15 g of m-hydroxybenzaldehyde, add 1.5 ml of nitroethane and 5 ml of absolute ethanol and dissolve under stirring, cool to -20°C, add 0.01 g of imidazole, 0.012 g of copper acetate monohydrate and 0.01 g of cinchonine, and Stir and react at -20∽-25°C for 35 hours until the reactant m-hydroxybenzaldehyde no longer decreases; adjust the pH value of the reaction solution to 3 with 20% hydrochloric acid under stirring, and spin evaporate the solvent under reduced pressure at 80°C After adding 10ml of water to dissolve the product, extract it three times with 3×10ml ethyl acetate, combine the ethyl acetate phases, and evaporate the solvent to dryness under reduced pressure at 50°C to obtain the crude product; after dissolving the crude product with 9ml dichloromethane, the After standing, the precipitated crystals were dried at 60°C to obtain 0.11 g ((R,S), (S,R) enantiomeric excess 75%) of the addition reaction product. Dissolve the crystals in 15ml of absolute etha...
Embodiment 2
[0043]Weigh 1.22g of m-hydroxybenzaldehyde, add 3ml of nitroethane and 40ml of absolute ethanol and dissolve it under stirring, cool down to -23°C, add 0.1g of imidazole, 0.07g of copper acetate monohydrate and 0.07g of cinchonine, in - Stir and react at 20∽-25°C for about 40 hours until the reactant m-hydroxybenzaldehyde is nearly complete; adjust the pH value of the reaction solution to 2 with 20% hydrochloric acid under stirring, and spin evaporate the solvent under reduced pressure at 80°C After adding 10ml of water to dissolve the product, use 4 × 25ml of ethyl acetate to extract 4 times, combine the ethyl acetate phases, and filter out after drying with anhydrous sodium sulfate overnight, and the solvent is evaporated to dryness under reduced pressure at 50°C to obtain the crude product; After the crude product was dissolved in 15ml of chloroform, it was placed at room temperature, and the precipitated crystals were dried at 60°C to obtain 0.76 g ((R,S), (S,R) enantiomeri...
Embodiment 3
[0046] Weigh 6.0 grams of m-hydroxybenzaldehyde, add 10 ml of nitroethane and 90 ml of absolute ethanol and dissolve it under stirring, cool to -20 ° C, add 0.5 grams of imidazole, 0.4 grams of copper acetate monohydrate and 0.4 grams of cinchonine, in - Stir and react at 15∽-20°C for 40 hours until the reactant m-hydroxybenzaldehyde no longer decreases; adjust the pH value of the reaction solution to 3 with 20% hydrochloric acid under stirring, and spin evaporate the solvent under reduced pressure at 80°C; After adding 50ml of water to dissolve the product, extract 3 times with 3×50ml of ethyl acetate, combine the ethyl acetate phases, and evaporate the solvent under reduced pressure at 50°C to obtain a crude product; after dissolving the crude product with 30ml of dichloroethane, After standing at room temperature, the precipitated crystals were dried at 60°C to obtain 4.3 g ((R,S), (S,R) enantiomeric excess 80%) of the addition reaction product. Dissolve the crystals in 60m...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More