Preparation method of precursor of ibrutinib
A system and system technology, applied in the field of preparation of pharmaceutical intermediates, can solve problems such as side reactions, low yields, and restrictions on industrial production, and achieve the effect of reducing the risk of side reactions
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
Embodiment 1
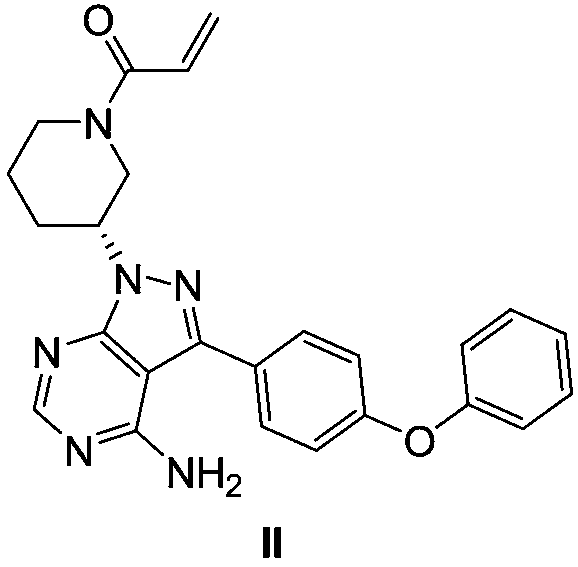
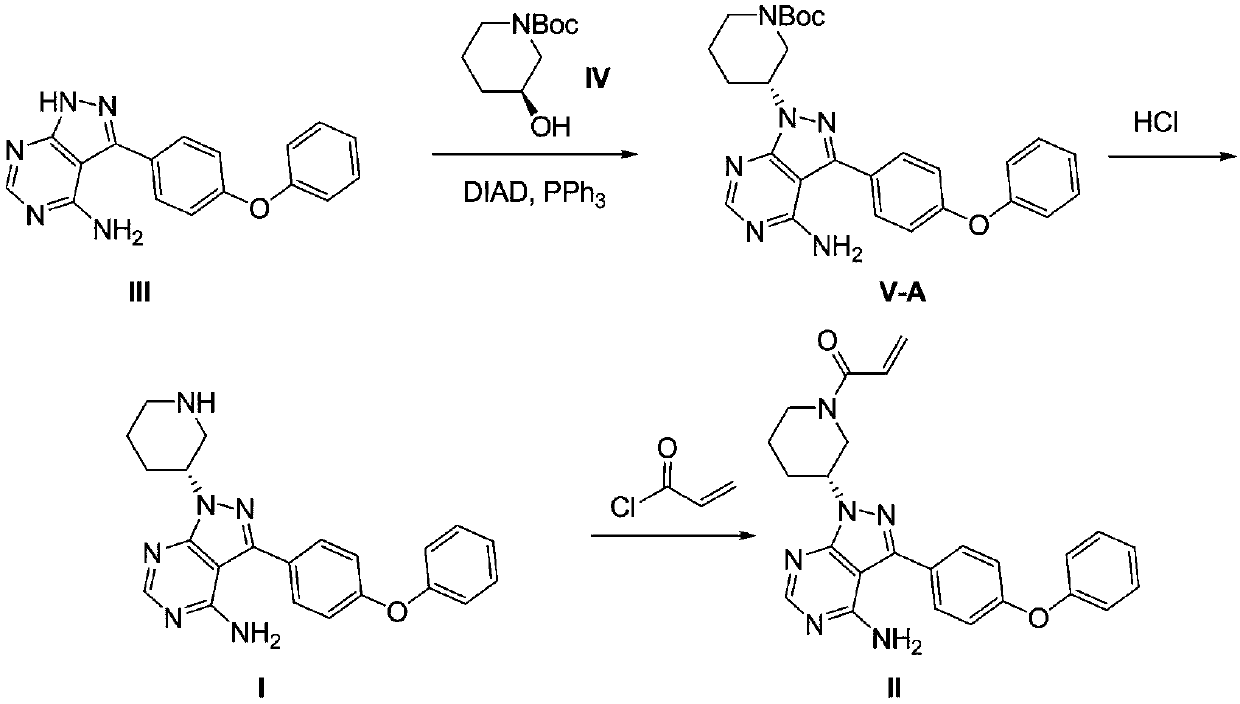
[0039] Example 1: Precursor (R)-3-(4-phenoxyphenyl)-1-(piperidin-3-yl)-1H-pyrazolo[3,4-d]pyrimidine Synthesis of -4-amine (I)
[0040]Add 100 g of compound (III), 350 g of triphenylphosphine and 2 L of tetrahydrofuran into the reaction flask. 270g of diisopropyl azodicarboxylate was added dropwise at 0°C. After dropping, the system was reacted at 50°C for 1 hour. HPLC showed that the content of compound (III) was less than 0.5%. Maintained at 50°C, 5 g of tetrabutylammonium bromide was added, followed by dropwise addition of 200 g of compound (IV) in 1 L of tetrahydrofuran. After 20 hours, keep the temperature at 50 degrees, add 250 mL of 30% hydrochloric acid solution dropwise, and stop the reaction after 2 hours. Cool down to room temperature, add 2.5 L of water to the system, and separate to obtain an aqueous layer. The aqueous layer was washed twice with 2 L of ethyl acetate each time. The liquid was separated to obtain the water layer, and solid sodium hydroxide was...
Embodiment 2
[0042] Example 2: (R)-3-(4-phenoxyphenyl)-1-(piperidin-3-yl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (Ⅰ )Synthesis
[0043] Add 100 g of compound (III), 260 g of triphenylphosphine and 1.8 L of tetrahydrofuran into the reaction flask. 173g of diethyl azodicarboxylate was added dropwise at 0°C. After dropping, the system was reacted at 55°C for 8 hours. HPLC showed that the content of compound (III) was less than 0.5%. Maintaining at 55°C, 11 g of tetrabutylphosphine bromide was added, followed by dropwise addition of a solution of 400 g of compound (IV) in 1.5 L of tetrahydrofuran. After 26 hours, keep the temperature at 55 degrees, add 400 mL of concentrated hydrochloric acid dropwise, and stop the reaction after 1.5 hours. Cool down to room temperature, add 3L of water to the system, and separate to obtain an aqueous layer. The aqueous layer was washed twice with 2 L of ethyl acetate each time. The liquid was separated to obtain the water layer, and solid sodium hydroxide ...
Embodiment 3
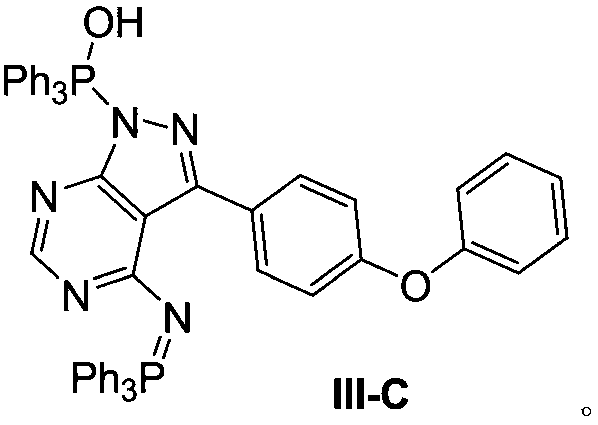
[0044] Embodiment three: the synthesis of compound (Ⅲ-C)
[0045] Add 1 g of compound (III), 2 g of triphenylphosphine and 10 mL of tetrahydrofuran into the reaction flask. Diisopropyl azodicarboxylate was added dropwise at 0°C. After dropping, the system was reacted at room temperature for 24 hours. HPLC showed that the content of compound (III) was less than 0.5%. Then 10 mL of water was added and stirred for another 24 hours. Concentrate under reduced pressure to remove the solvent, and the resulting oil is separated by column chromatography to obtain 2g of the oil:
[0046] 1H NMR (500MHz, CDCl3) δ: 7.12-7.15(m, 5H), 7.37-7.42(m, 8H), 7.44-7.47(m, 8H), 7.50-7.55(m, 7H), 7.68-7.72(m , 7H), 7.83-7.87 (m, 5H), 8.19 (s, 1H), 8.53 (d, J=8.5Hz, 2H).
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More - R&D
- Intellectual Property
- Life Sciences
- Materials
- Tech Scout
- Unparalleled Data Quality
- Higher Quality Content
- 60% Fewer Hallucinations
Browse by: Latest US Patents, China's latest patents, Technical Efficacy Thesaurus, Application Domain, Technology Topic, Popular Technical Reports.
© 2025 PatSnap. All rights reserved.Legal|Privacy policy|Modern Slavery Act Transparency Statement|Sitemap|About US| Contact US: help@patsnap.com