

A Single-Sample Genome-Wide Method for Predicting Allele-Specific Copy Number Variations
An allele-specific and copy number variation technology, applied in the field of bioinformatics, can solve the problems of high sequencing costs, high false negatives, and increased costs, and achieve the effect of low sequencing depth and high detection accuracy
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
Embodiment 1
[0136] In this embodiment, a tumor sample refers to a tumor sample, and a normal sample refers to a normal sample.
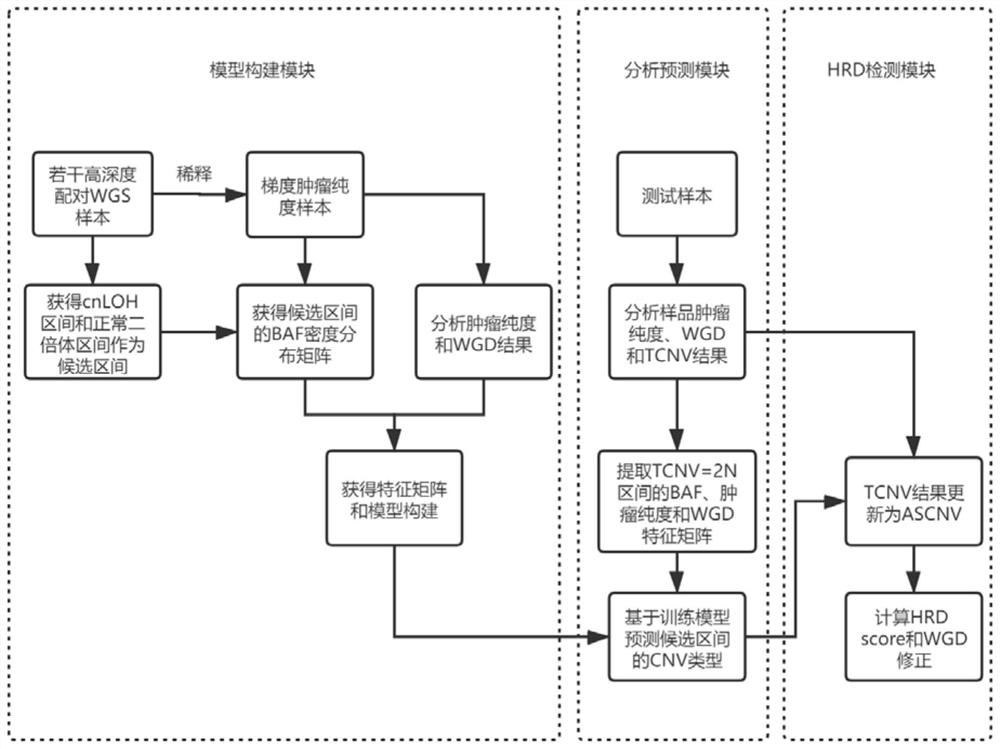
[0137] Such as figure 1 As shown, the steps of each module in this embodiment are as follows:
[0138] 1. Model building blocks
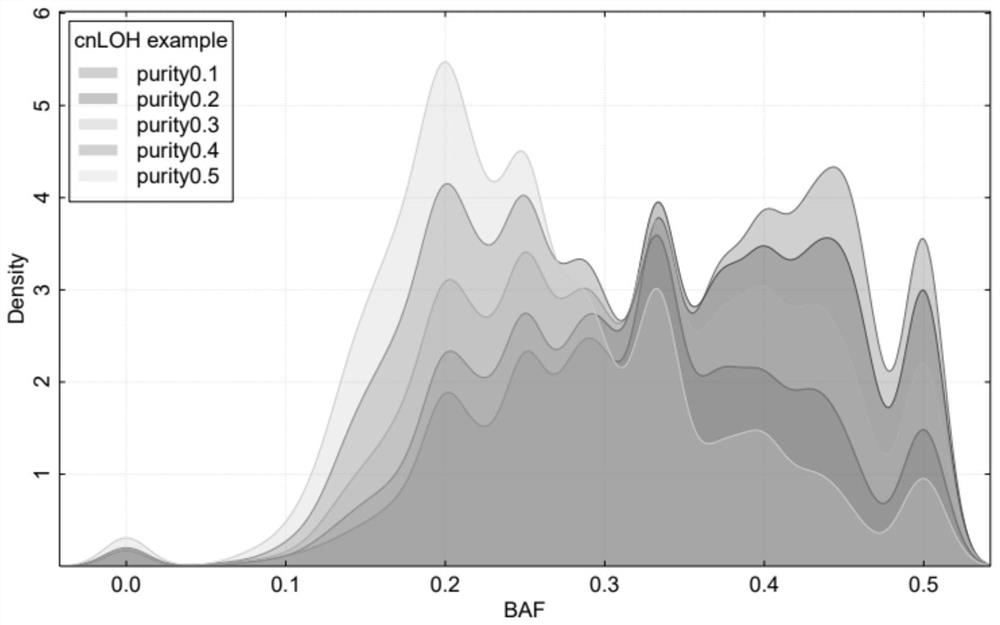
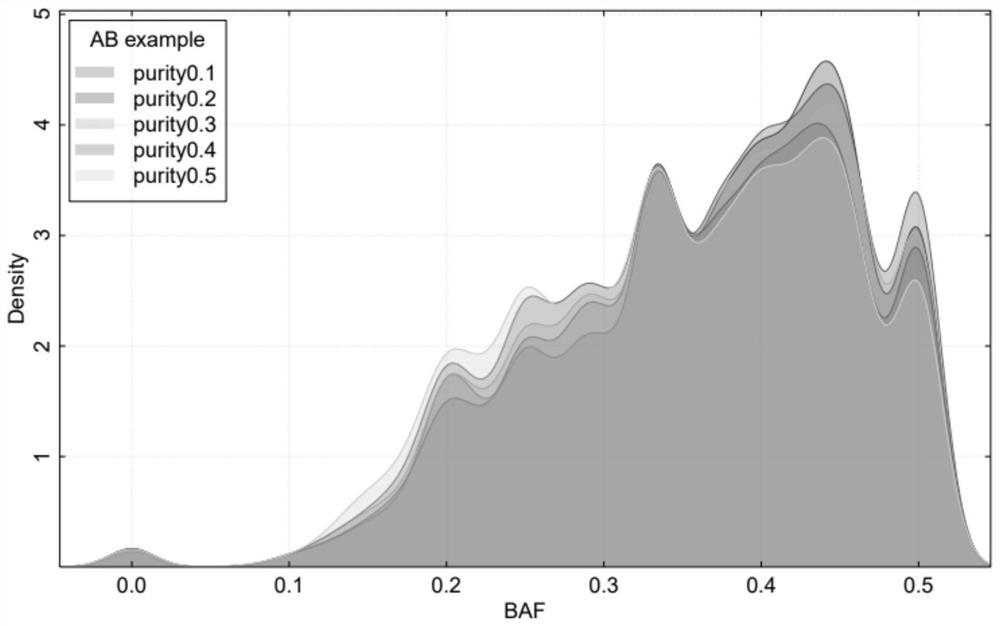
[0139]In this embodiment, 148 cases of paired tumor samples (that is, each tumor sample is paired with a normal sample from the same individual, the tumor sample is a tumor tissue sample, and the normal sample is a paracancerous tissue sample) high-depth whole-genome sequencing data (the sequencing depth is 30×). It covers healthy people (the tumors of healthy people are nodules or benign tumors) and the four major types of cancer (ovarian cancer, breast cancer, prostate cancer, bladder cancer). Data quality filtering (Q20>80%, N<5%); use BWA software to compare to the human reference genome hg19, evaluate the contamination rate of the sample, and remove samples with a high contamination rate (specifically remove samples with a com...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More