

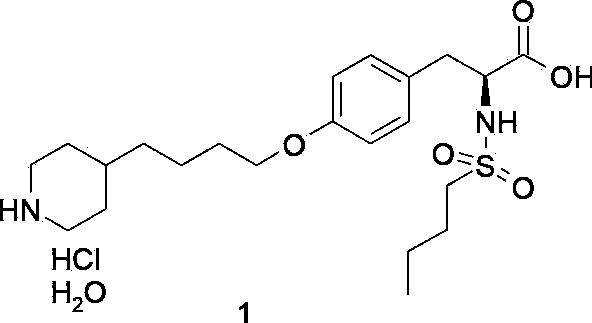
Method of preparing tirofiban hydrochloride
A technology for tirofiban and hydrochloric acid, which is applied in the field of preparation of tirofiban hydrochloride, can solve the problems of high risk and high equipment requirements, and achieve the effects of low production equipment requirements, simple synthesis, and safe production
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
Embodiment 1
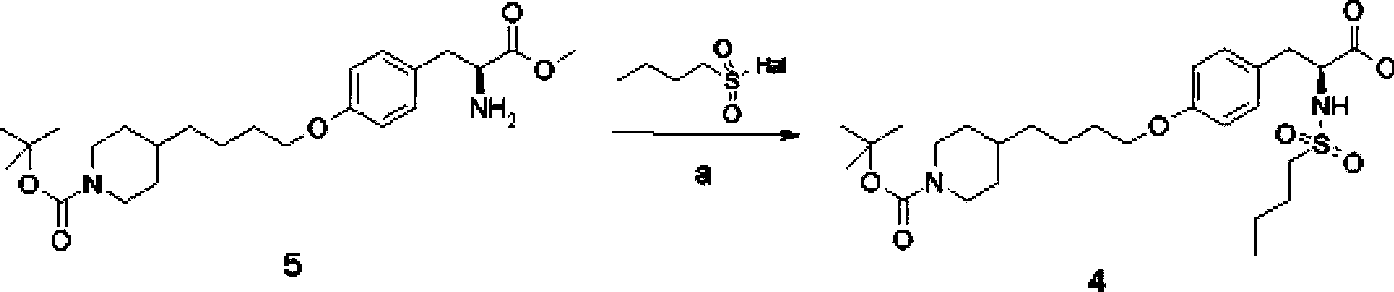
[0075] Embodiment 1: the preparation of compound 4
[0076] Compound 5 (Shanghai Fengyi Chemical Technology Co., Ltd.) (80 g) was dissolved in 800 mL of acetonitrile, and 600 mL of water was added. After cooling in an ice bath to below 5°C, disodium hydrogen phosphate (78.5 g) was added in portions. Control the temperature at 5-10°C, add n-butylsulfonyl chloride (35.3 mL) dropwise, and drop it in about 5 minutes. Remove the ice bath and let it rise to room temperature (17°C) naturally. After insulated and stirred for 8 hours, HPLC monitored that the reaction was complete, and the reaction was stopped. Stand to separate and separate, the lower aqueous phase was extracted with methyl tert-butyl ether (500mL); the upper organic phase was rotary evaporated under reduced pressure to remove acetonitrile, the obtained oil was dissolved in 800mL methyl tert-butyl ether, and combined with the previous extract . The resulting solution was washed with water (1L), 0.25N hydrochloric ...
Embodiment 2
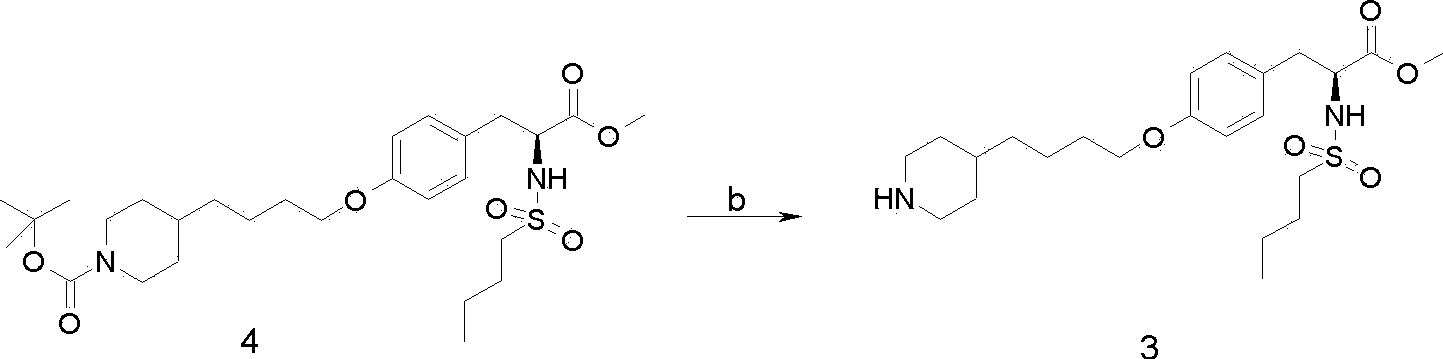
[0077] Embodiment 2: the preparation of compound 3
[0078] The crude compound 4 (112 g) obtained in Example 1 was dissolved in 480 mL of dichloromethane, and cooled to 5° C. in an ice bath. The temperature was controlled below 10°C, and trifluoroacetic acid (93 mL) was added dropwise, and the dropwise was completed in about 15 minutes. The ice bath was removed, the temperature was naturally raised to 15° C., and the reaction was carried out with heat preservation and stirring for 6 hours. The completion of the reaction was monitored by HPLC, and water (1 L) and dichloromethane (300 mL) were added to the reaction solution, stirred for 10 minutes, and allowed to stand to separate into layers. The lower organic phase was washed with water (1L*2). Dichloromethane was distilled off under reduced pressure to obtain a pale yellow oil. Then the obtained oil was dissolved in methanol (500 mL), 1 L of water, n-hexane (500 mL), and methyl tert-butyl ether (500 mL) were added, stirred...
Embodiment 3
[0079] Embodiment 3: the preparation of compound 2
[0080] Compound 3 (80 g) obtained in Example 2 was dissolved in 250 mL of methanol, 250 mL of water was added, and cooled to below 5° C. in an ice bath. Protected under nitrogen and protected from light, add lithium hydroxide aqueous solution (20.77g lithium hydroxide dissolved in 250mL water) dropwise, and finish dropping in about 20 minutes, keeping the temperature below 10°C. The ice bath was removed, and the temperature was raised to 15°C naturally. After insulated and stirred for 3.5 hours, the reaction was stopped. The reaction solution was cooled to 8°C, neutralized by adding 35% acetic acid aqueous solution (350mL) dropwise, and the dropwise was completed in 20 minutes, and the temperature was controlled below 15°C. Keep stirring for 15 minutes, then steam under reduced pressure (30°C) for about 45 minutes. Then the reaction solution was heated to 75°C, stirred for 30 minutes, and then removed from the heat. Natu...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More