

Method for preparing tribenoside
A technology of tribenzyl glucoside and tribenzyloxy group, which is applied in the field of preparation of tribenzyl glucoside, can solve the problems of low purity and long production cycle, and achieve the effect of good quality and high purity of the final product and avoid column chromatography operation
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

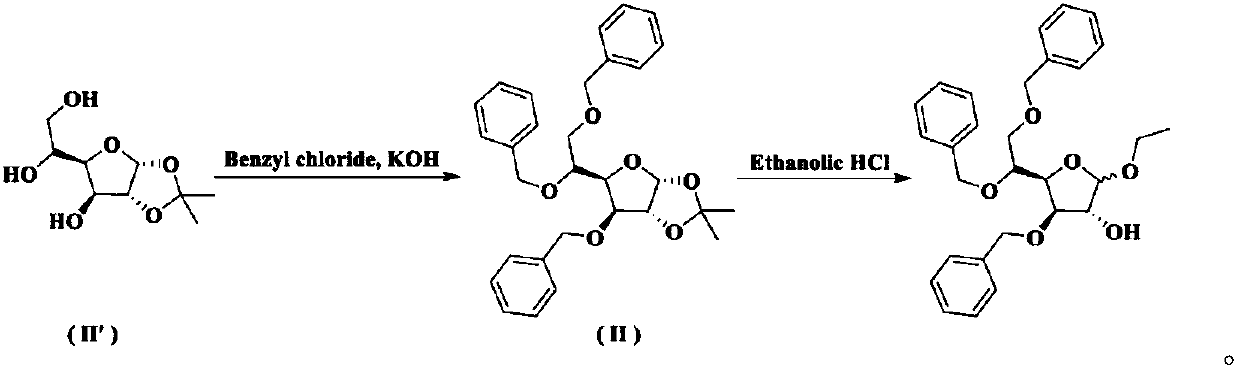
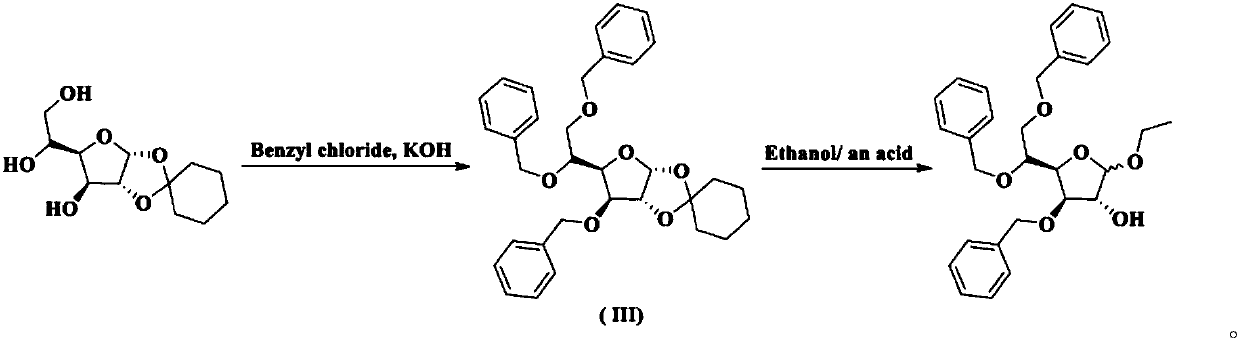
Examples
Embodiment 1
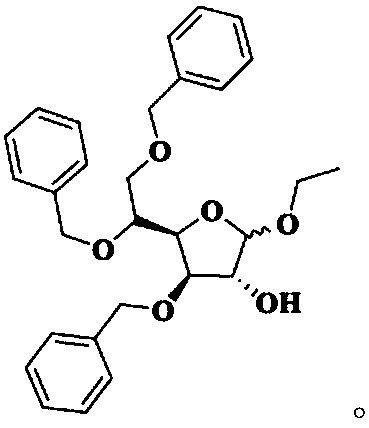
[0035] Synthesis of Intermediate I
[0036] At room temperature, add 8.40 kg of purified water into the three-necked bottle, slowly add 0.22 kg of concentrated sulfuric acid under stirring, and after stirring evenly, add 1.10 kg of acetic acid. When the temperature of the reaction solution rises to 65°C, add 1.10kg of 3,5,6-tribenzyloxy-1,2-oxo-isopropylidene-α-D-glucofuranose at one time, and then control the temperature at 65°C for reaction 10min. Under stirring, pour 2.60 kg of crushed ice into the reaction solution, add 10.60 L of dichloromethane, stir for 30 minutes, separate the organic phase; extract the water phase with 2.30 L of dichloromethane, combine the organic phases and wash with 10.60 × 2 kg of purified water; The dichloromethane phase was washed with a pre-prepared solution of 0.12kg sodium bicarbonate and 8.60kg purified water to pH=7~8; the dichloromethane phase was washed with 10.60kg purified water, and 3.00kg anhydrous sulfuric acid was added to the dich...
Embodiment 2
[0038] Synthesis of Intermediate I
[0039] At room temperature, add 8.40 kg of purified water into the three-necked bottle, slowly add 5.5 kg of concentrated sulfuric acid under stirring, and after stirring evenly, add 13.2 kg of acetic acid. When the temperature of the reaction solution rises to 100°C, add 1.10kg of 3,5,6-tribenzyloxy-1,2-oxo-isopropylidene-α-D-glucofuranose at one time, and then control the temperature at 100°C for reaction 120min. Under stirring, pour 2.60 kg of crushed ice into the reaction solution, add 10.60 L of dichloromethane, stir for 30 minutes, separate the organic phase; extract the water phase with 2.30 L of dichloromethane, combine the organic phases and wash with 10.60 × 2 kg of purified water; The dichloromethane phase was washed with a pre-prepared solution of 1.2kg sodium bicarbonate and 8.60kg purified water to pH=7~8; the dichloromethane phase was washed with 10.60kg purified water, and 3.00kg anhydrous sulfuric acid was added to the dic...
Embodiment 3
[0041] Synthesis of Intermediate I
[0042] At room temperature, add 8.40kg of purified water into the three-necked bottle, slowly add 0.22kg of concentrated sulfuric acid under stirring, and after stirring evenly, add 1.1kg of formic acid. When the temperature of the reaction solution rises to 65°C, add 1.10kg of 3,5,6-tribenzyloxy-1,2-oxo-isopropylidene-α-D-glucofuranose at one time, and then control the temperature at 65°C for reaction 10min. Under stirring, pour 2.60 kg of crushed ice into the reaction solution, add 10.60 L of dichloromethane, stir for 30 minutes, separate the organic phase; extract the water phase with 2.30 L of dichloromethane, combine the organic phases and wash with 10.60 × 2 kg of purified water; The dichloromethane phase was washed with a pre-prepared solution of 0.12kg sodium bicarbonate and 8.60kg purified water to pH=7~8; the dichloromethane phase was washed with 10.60kg purified water, and 3.00kg anhydrous sulfuric acid was added to the dichloro...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More