

Synthesis method of clofarabine of nucleoside analogues
A compound, hydrobromic acid technology, applied in the directions of drug combination, sugar derivatives, organic chemistry, etc., can solve the problems of complicated operation steps, expensive reagents, etc., and achieve the effects of high yield, easy operation and mild conditions
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

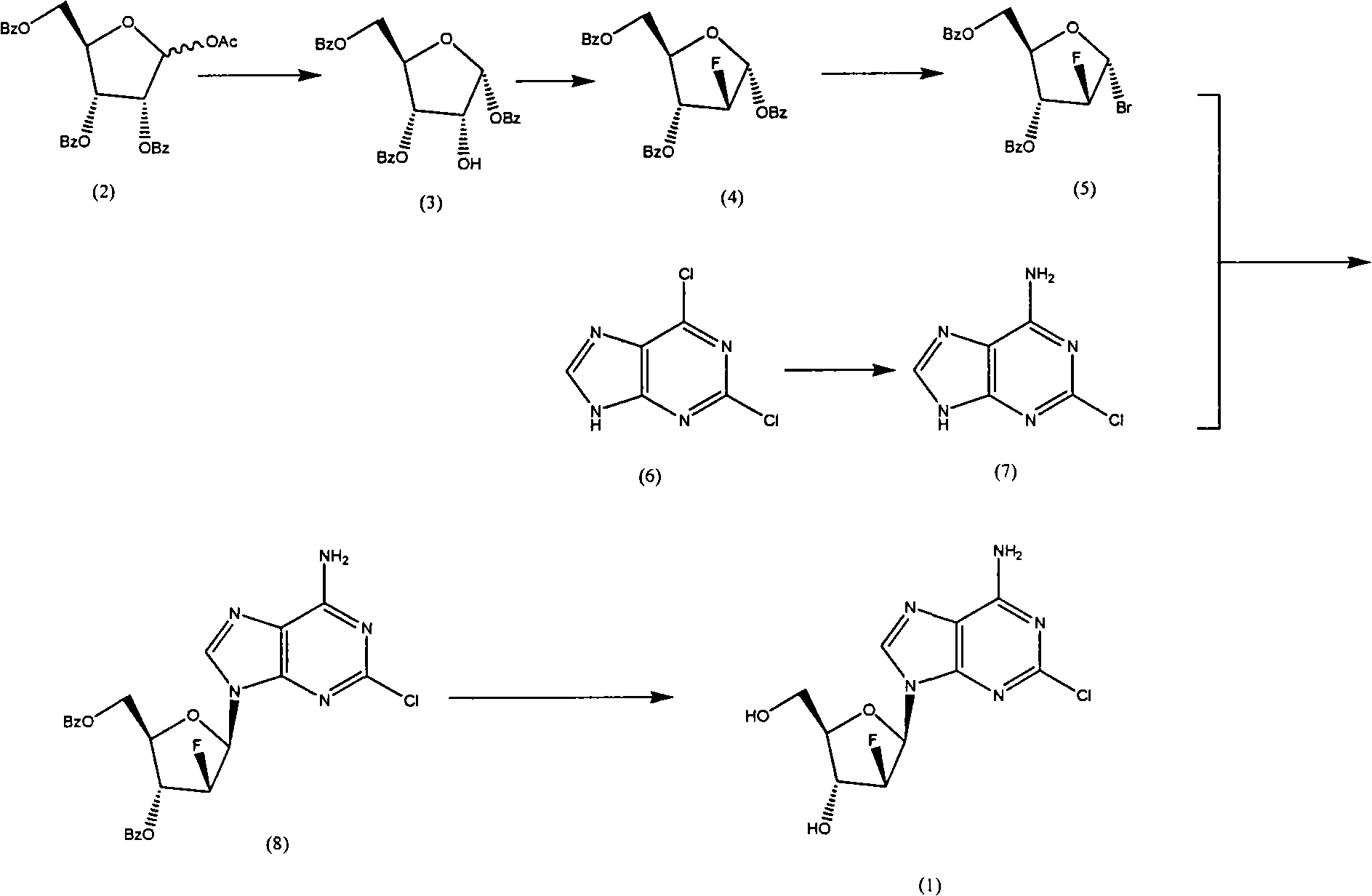
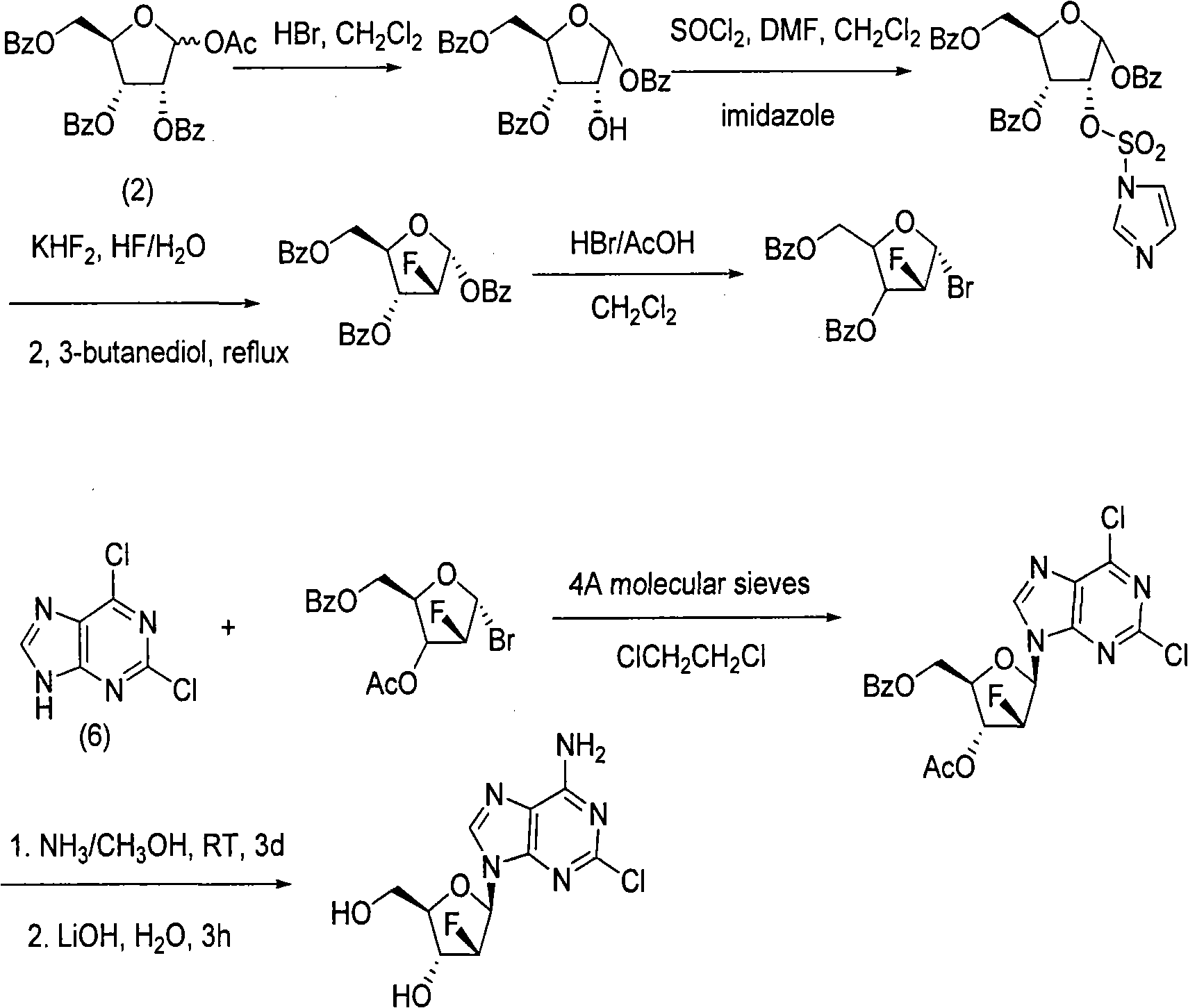
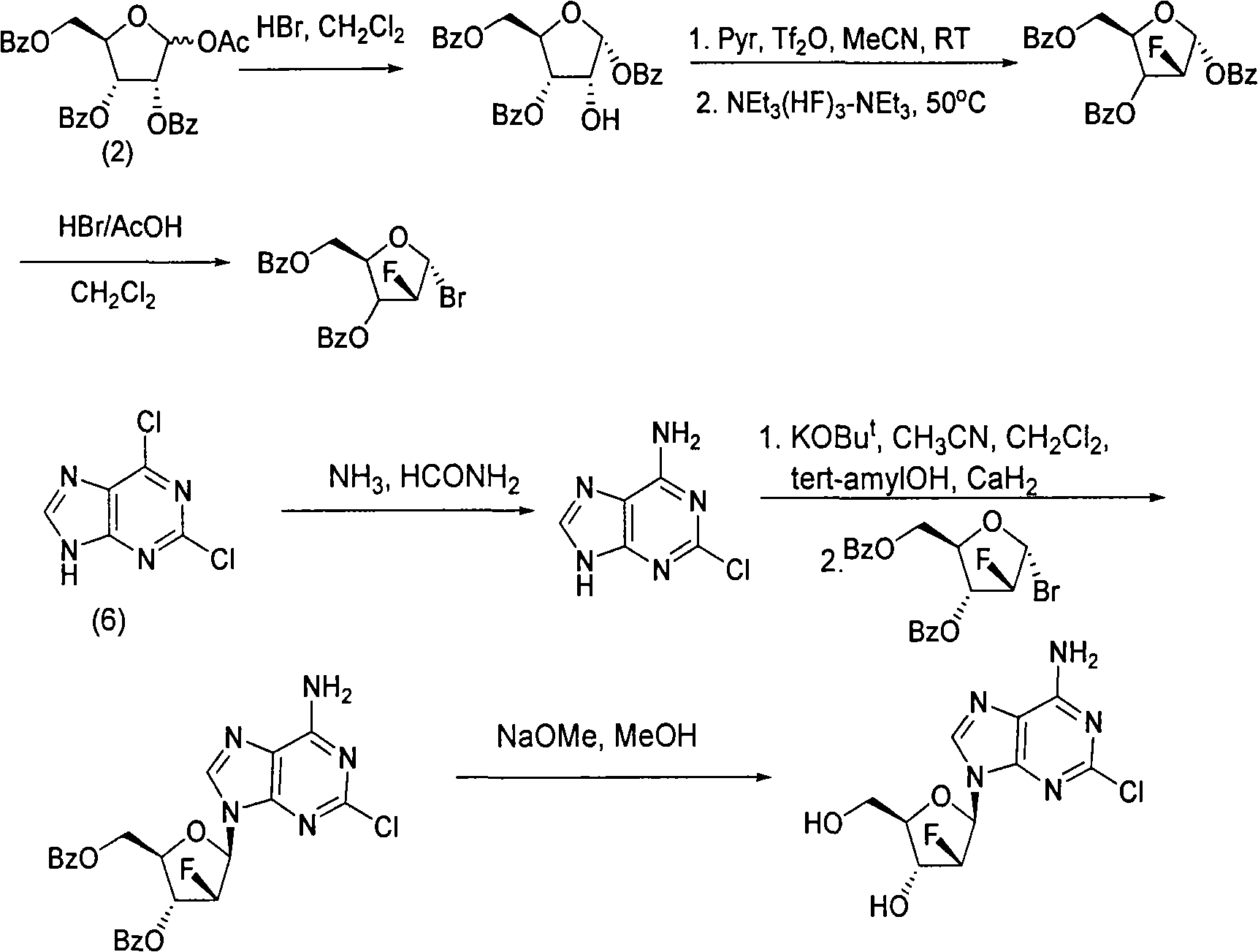
Examples
Embodiment 1
[0043] Preparation of compound (3): synthesis of 1,3,5-tris-oxy-benzoyl ribose (3)
[0044] Add 500mL of dichloromethane and 9.7mL of methanol to a 1L reaction flask, cool to 0~5℃ in an ice-water bath, carefully add 17.8mL of acetyl bromide while stirring, stir for 10 minutes at 0℃, add 100.9g (1), keep the temperature React at 0℃~5℃ for about 2 hours. After the reaction is complete, add 200 mL of water, stir at room temperature for 1 hour, separate the organic layer, extract the aqueous layer with dichloromethane (2×200 mL), combine the organic layers, and dry with magnesium sulfate About half an hour, filter, concentrate to about 300mL, cool to 0°C in an ice-water bath, add 600mL n-heptane while stirring, a white solid precipitates, continue stirring at 0°C for 2 hours, filter, the crystals are 3×50mL n-heptane / two Wash with methyl chloride (2 / 1), immediately transfer the crystals to a beaker, and put them in a vacuum desiccator to drain. 42 g of white powder (2) was obtained, w...
Embodiment 2
[0046] Preparation of compound (4): Synthesis of 2-deoxy-2-fluoro-1,3,5-tri-oxy-benzoyl-α-D-ribofuranose (4)
[0047] Add 13.9g (2) and 200mL dichloromethane into a 500mL reaction flask, add 14.5g DAST under stirring, heat to 40~42℃ and stir for about 18 hours, cool to room temperature, add saturated NaHCO 3 200mL of the solution, stirred for about 30 minutes, transferred to a separatory funnel, separated the organic phase, and then washed the organic phase with water (2×200mL), dried with magnesium sulfate for about half an hour, filtered, evaporated the solvent under reduced pressure, and the residue was diluted to 100ml Recrystallize from absolute ethanol. 9.2 g of white powder (4) was obtained, with a yield of 66%. 32g of compound (3) (195mmol) was added to 350ml of DMF (anhydrous treatment), 28g of sodium hydride was added under ice-water bath, stirred for half an hour, 97ml of 2,4-dichlorobenzyl chloride was added dropwise, and the reaction was carried out at 35°C. After 4 h...
Embodiment 3
[0049] Preparation of compound (5): Synthesis of 1-bromo-2-deoxy-2-fluoro-3,5-di-oxy-benzoyl-α-D-ribofuranose (5)
[0050] Add 13.8g (4) and 70mL dichloromethane to a 250mL reaction flask, add 17.5mL hydrogen bromide acetic acid solution (33%) under stirring at room temperature, stir at room temperature for about 18 hours, and then water (2×200mL), saturated NaHCO 3 The organic phase was washed with 200 mL of the solution, dried with magnesium sulfate, filtered, and the solvent was evaporated under reduced pressure to obtain 12.4 g of oil (4) with a yield of 99%
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More