

Catalyst containing trace noble metals for dehydrogenating organic hydrogen storage medium and preparation method
A catalyst and precious metal technology, applied in the field of composite catalyst and preparation of organic liquid hydrogen storage medium dehydrogenation, can solve the problem of low selectivity, and achieve the effects of improving catalytic activity, reducing cost and reducing dosage
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

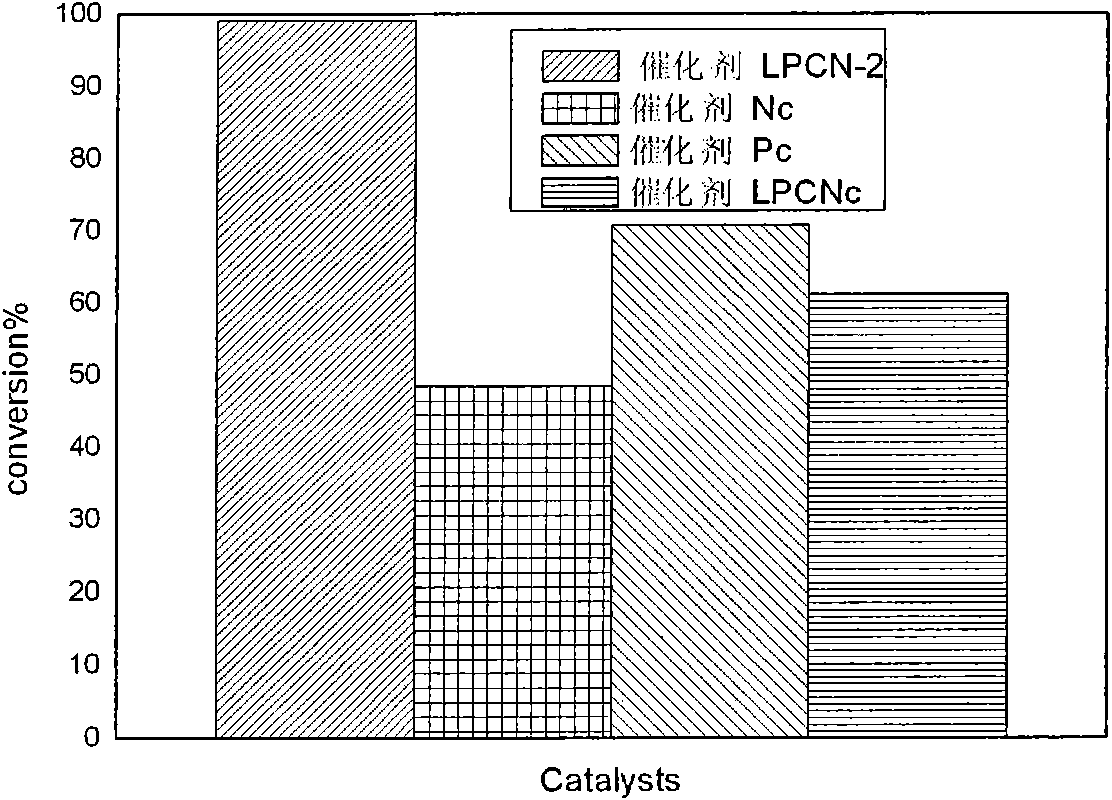
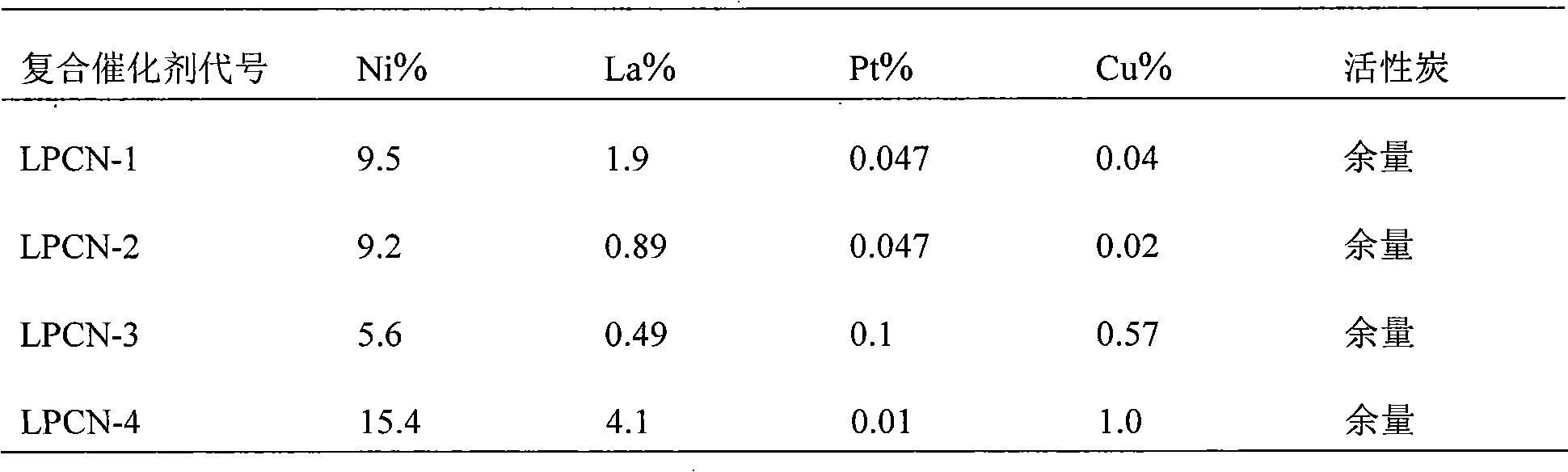

Examples
Embodiment 1
[0035] a. 2.9g nickel acetate Ni(AC) 2 .4H 2 O and 0.43g lanthanum nitrate La(NO 3 ) 3 .6H 2 O was dissolved in 4.0ml of deionized water, 2.0ml of ethanol and 1.0ml of glacial acetic acid; then weighed 6.0g of activated carbon desorbed under vacuum at 120°C and added to the above mixed solution. The above mixture was dispersed under ultrasound for at least 20 minutes, left to stand for 48 hours, and then dried with a rotary evaporator, calcined at 400°C for 3 hours under nitrogen protection, and cooled to room temperature to obtain the catalyst precursor A1.
[0036] b. With 0.012g copper nitrate Cu(NO 3 ) 2 .3H 2 O and 0.60g of activated carbon were dissolved in 4.0ml of deionized water, 2.0ml of ethanol and 1.0ml of glacial acetic acid; the same ultrasonic dispersion, evaporative drying and calcination process as in step a was adopted, wherein the calcination temperature was 380°C and cooled to room temperature to obtain Cu-containing catalyst precursor B1.
[0037] ...
Embodiment 2
[0043] a. 2.8g nickel acetate Ni(AC) 2 .4H 2 O and 0.20g lanthanum nitrate La(NO 3 ) 3 .6H 2 O was dissolved in 4.0ml of deionized water, 2.0ml of ethanol and 0.8ml of glacial acetic acid; then weighed 6.0g of activated carbon after vacuum desorption and added to the above mixed solution. The above mixture was left to stand for 48 hours, dried with a rotary evaporator, calcined at 400° C. for 3 hours under nitrogen protection, and cooled to room temperature to obtain catalyst precursor A2.
[0044] b. With 0.0060g copper nitrate Cu(NO 3 ) 2 .3H 2 O and 0.60g of activated carbon were dissolved in 4.0ml of deionized water, 2.0ml of ethanol and 1.0ml of glacial acetic acid; using the same ultrasonic dispersion, evaporative drying and calcination process as in step a, wherein the calcination temperature was 400°C and cooled to room temperature to obtain Cu-containing catalyst precursor B2.
[0045] c. Mix the precursors A2 and B2, and then use the temperature-programmed re...
Embodiment 3
[0050] a. 1.64g nickel chloride NiCl 2 .6H 2 O and 0.09 g lanthanum chloride LaCl 3 .6H 2 O was dissolved in 4.0ml of deionized water, 2.0ml of ethanol and 0.8ml of glacial acetic acid; then weighed 6.0g of activated carbon after vacuum desorption and added to the above mixed solution. The above mixture was dispersed under ultrasound for 30 min, left to stand for 48 hours, dried with a rotary evaporator, calcined at 400° C. for 3 hours under nitrogen protection, and cooled to room temperature to obtain catalyst precursor A3.
[0051] b. With 0.11g copper chloride CuCl 2 .2H 2 O and 0.60g of activated carbon were dissolved in 4.0ml of deionized water, 2.0ml of ethanol and 1.0ml of glacial acetic acid; the same ultrasonic dispersion, evaporative drying and calcination process as in step a was adopted, wherein the calcination temperature was 450°C and cooled to room temperature to obtain Cu-containing catalyst precursor B3.
[0052] c. Mix the precursors A3 and B3, and then...
PUM
| Property | Measurement | Unit |
|---|---|---|
| specific surface area | aaaaa | aaaaa |
Abstract
Description
Claims
Application Information
 Login to View More
Login to View More