

Method for preparing fondaparinux sodium intermediate
A technology for fondaparinux sodium and intermediates, which is applied in the field of preparation of intermediates, can solve problems such as restricting large-scale use, forming bottlenecks in the production stage, and complex synthesis of fondaparinux.
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

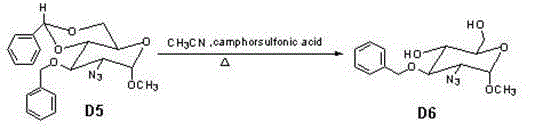
Examples
Embodiment 1
[0042] Example 1 Preparation of Compound D1
[0043] In a 2L round bottom flask, add 100g of PVP-DVB as the carrier of sulfonic acid H + The cation exchange resin was washed 4 times with methanol solution until the pH value of the methanol solution reached neutrality. Then add 800ml of methanol and 100g of N-acetylglucosamine. According to the reaction conditions in Table 1, the reflux reaction was maintained. After the reaction, filter the resin immediately after cooling to 45°C, adjust the pH of the filtrate to 7-9, and distill under reduced pressure. Recrystallize with toluene and dry under vacuum at 45°C to obtain the finished product D1. H-NMR: 7.5-8.5 (d, 1H, NH), 3.4-5.5 (m, 5H, 5×CH), 3.5-3.8 (d, 2H, O-CH 2 ), 3.3 (s, 3H, O-CH 3 ), 2.1 (s, 3H, CH 3 ), 1.5-4.0 (m, 3H, 3×OH). m / z: 235.11.
[0044] Table 1 Reaction conditions
[0045] temperature reflex Reaction time yield 1 50℃ 10h 71% 2 60℃ 40h 80% 3 70℃ 70h 78%
Embodiment 2
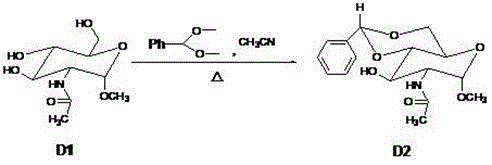
[0046] Example 2 Preparation of Compound D2
[0047]According to the reaction conditions in Table 2, compound D1, acetonitrile and benzaldehyde dimethyl acetal were put into a 500ml round bottom flask. When D1 is completely dissolved, add p-toluenesulfonic acid. Raise the temperature to reflux the reaction solution. After the reaction is complete, cool to room temperature and adjust the pH of the reaction solution to 7-9. The crude product was obtained by vacuum distillation, recrystallized with n-hexane, and finally unreacted D1 was removed with water, and the product D2 was obtained by vacuum drying at 45°C. H-NMR: 7.5-8.5 (d, 1H, NH), 7.0-7.3 (m, 5H, 5×Ar-H), 5.9-6 (s, 1H, Ph-CH), 3.0-5.5 (m, 5H , 5×CH), 3.7-4.1(d, 2H, O-CH 2 ), 3.3 (s, 3H, O-CH 3 ), 2.1 (s, 3H, CH 3 ), 1.5-4.0 (m, 1H, OH). m / z: 323.2.
[0048] Table 2 Reaction conditions
[0049] Compound D1 Acetonitrile Benzaldehyde dimethyl acetal p-Toluenesulfonic acid temperature reflex ...
Embodiment 3
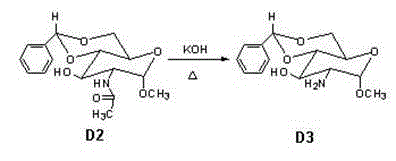
[0050] Example 3 Preparation of Compound D3
[0051] According to the reaction conditions in Table 3, 16 g of compound D2 was put into a 1000 ml round bottom flask. Dissolve 34.7g KOH in 600ml water, and add the obtained potassium hydroxide solution into the round bottom flask. After the addition was complete, the temperature was raised and the reaction was refluxed. After the reaction is complete, cool to 0~5°C and stir for 10h. After suction filtration, the crystals were first washed with water and then with methyl tert-butyl ether, and dried in vacuum at 45° C. for 12 hours to obtain product D3. H-NMR: 7.0-7.3(m, 5H, 5×Ar-H), 5.9-6(s, 1H, Ph-CH), 3.0-5.5(m, 5H, 5×CH), 3.7-4.1(d , 2H, O-CH 2 ), 3.3 (s, 3H, O-CH 3 ), 1-3(d,2H,NH 2 ), 1.5-4.0 (m, 1H, OH). m / z: 281.1.
[0052] Table 3 Reaction conditions
[0053] temperature reflex Reaction time yield 1 60℃ 20h 59% 2 70℃ 45h 70% 3 80℃ 70h 63%
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More