

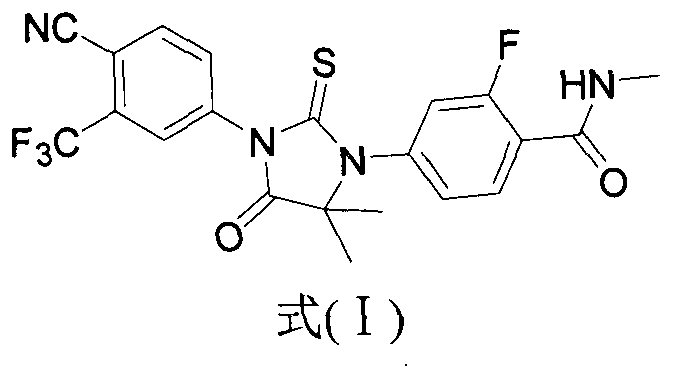
Method for synthesizing enzalutamide
A synthetic method, enzalutamide technology, applied in the field of enzalutamide synthesis, can solve the problems of low overall yield and high toxicity, and achieve the effects of simple operation, reduced environmental and human hazards, and reduced raw material costs
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
Embodiment 1
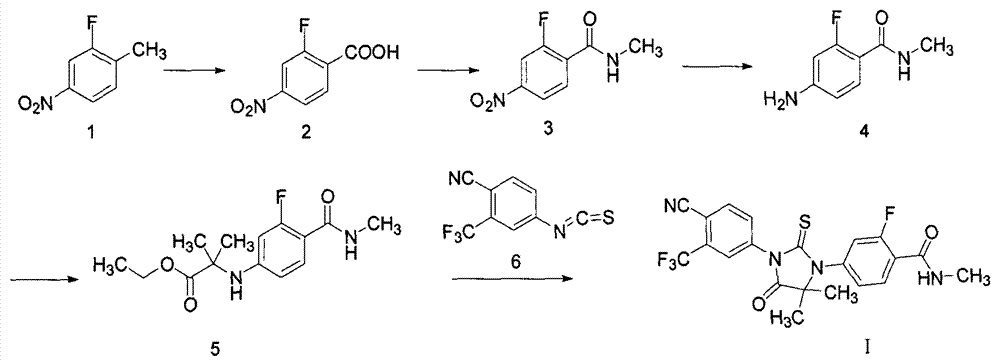
[0031] Add compound 1 (15.5g, 0.10mol) and 200mL water into a 500mL three-necked flask, add potassium permanganate (36.4g, 0.23mol) in batches, and then reflux for 8h. TLC detected that the reaction was complete, and after the purple color of the reaction solution faded, it was suction filtered while it was hot. If the purple color of the reaction solution does not fade, add an appropriate amount of saturated sodium bisulfite solution to completely fade the reaction solution. The filtrate was cooled with an ice-water bath, and the pH was adjusted to 1 with concentrated hydrochloric acid. The solvent was evaporated under reduced pressure. Recrystallization from water gave compound 2 (10.2 g, 55%) as white needle crystals.
Embodiment 2
[0033] Add compound 1 (15.5g, 0.10mol), 100ml acetic acid, 50ml sulfuric acid into a 500mL three-necked flask, add chromium trioxide-acetic anhydride (30.3g, 0.15mol) in batches, then raise the temperature to 50°C for 5h. It was detected by TLC that the reaction was complete. 300 ml of water was added to the reaction solution, extracted three times with 150 ml of dichloromethane, washed three times with 150 ml of saturated sodium chloride, and the organic layer was dried with anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure. Column chromatography yielded compound 2 (10.2 g, 63%) as white needle crystals. 1 H NMR (300MHz, CDCl 3 )δ: 10.59 (s, 1H), 8.28-8.05 (m, 3H); EI-MS m / Z: 83, 131, 185 [M] + .
[0034] Synthesis of compound 3
Embodiment 3
[0036] Compound 2 (3.7g, 0.02mol), 50mL DMF was added to a 250mL three-necked flask, and thionyl chloride (9.5g, 0.08mol) was slowly added dropwise to the reaction solution under nitrogen protection, and reacted at room temperature for 12h, then, 40 Evaporate excess thionyl chloride under reduced pressure at ℃, pass methylamine gas into the reaction solution, control the temperature below 30 ℃, stop when the reaction solution is detected to be neutral. Extracted three times with 300ml ethyl acetate, washed three times with 150ml saturated sodium chloride, and dried the organic layer with anhydrous magnesium sulfate. The solvent was distilled off under reduced pressure, and recrystallized from ethanol to obtain compound 3 (3.6 g, 91%) as a yellow solid. 1 H NMR (300MHz, CDCl 3 )δ: 8.33-8.31(m, 1H), 8.13(d, J=2.7Hz, 1H), 8.04(d, J=8.3Hz, 1H), 6.75(s, 1H), 3.09(s, 3H); EI-MS m / Z: 94, 122, 168, 198 [M] + .
[0037] Synthesis of Compound 4
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More