

Synthesis method of Velpatasvir intermediate A
A synthetic method, the technology of velpatasvir, applied in the field of medicine, can solve the problems of restricting industrial application, low purity of velpatasvir intermediate A, high synthesis cost of velpatasvir intermediate A, and achieve good application prospects and Market potential, beneficial to industrial production, mild reaction conditions
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

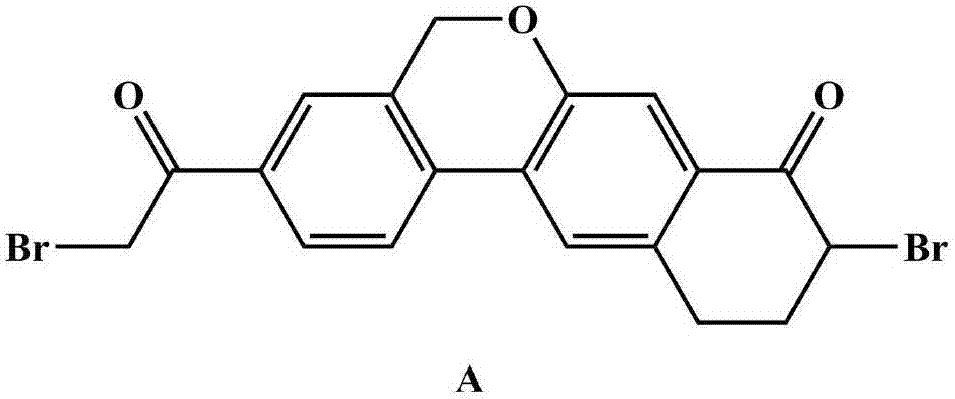
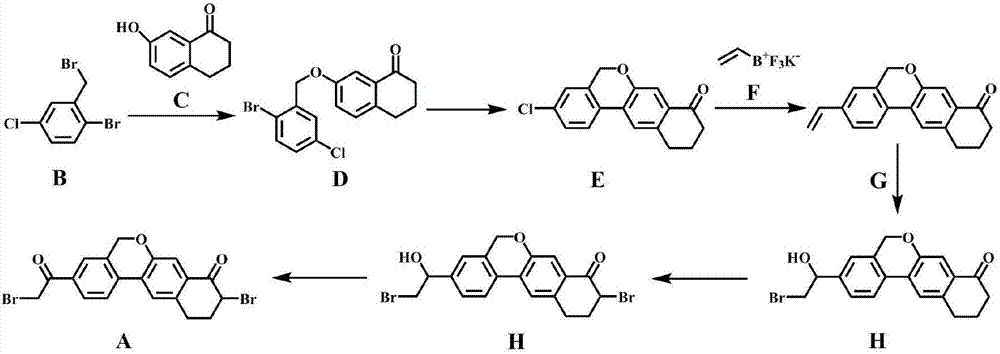
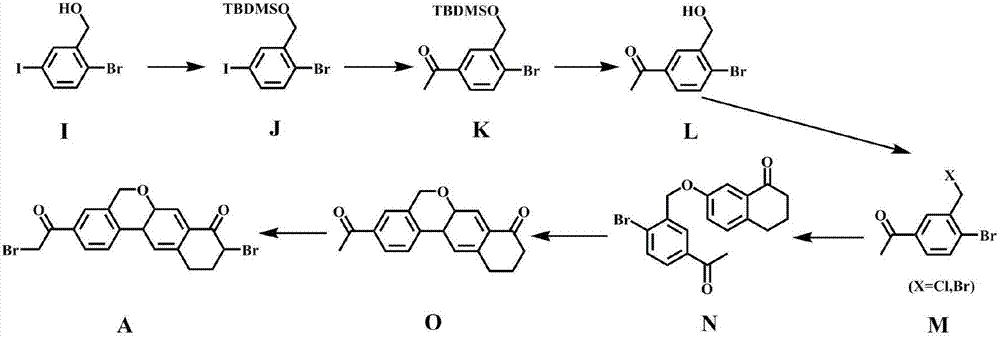
Examples
Embodiment 1
[0073] Under nitrogen protection, 40.0 g of compound 4, 28.0 g of triethylamine and 400.0 mL of dichloromethane were added to a 1 L reaction flask. After cooling down to 0°C, 15.0 g of acetyl chloride was added dropwise. After the dropwise addition, the reaction solution was raised to room temperature, and stirred for 2 hours.
[0074] TLC detected that the reaction was complete, and 200.0 mL of water was added to the reaction bottle, and after stirring for 0.5 hour, liquid separation was carried out to obtain an organic phase. The organic phase was washed once with 200 mL of saturated brine, and concentrated to dryness at 50°C under reduced pressure to obtain 44.0 g of compound 5 as a pale yellow oil with a purity of 95.2% and a yield of 92.9%.
[0075] MS calcd for C 11 h 11 BrO 3 Na(M+Na) + :294.1, found: 294.1.
Embodiment 2
[0077] Under nitrogen protection, 40.0 g of compound 4, 28.0 g of triethylamine and 400.0 mL of dichloromethane were added to a 1 L reaction flask. After cooling down to 0°C, 15.0 g of acetyl chloride was added dropwise. After the dropwise addition was completed, the reaction solution was raised to 40° C. and stirred for 30 minutes.
[0078] TLC detected that the reaction was complete, and 200.0 mL of water was added to the reaction bottle, and after stirring for 0.5 hour, liquid separation was carried out to obtain an organic phase. The organic phase was washed once with 200 mL of saturated brine, and concentrated to dryness at 50°C under reduced pressure to obtain 40.7 g of compound 5 as a pale yellow oil with a purity of 91.4% and a yield of 86.0%.
[0079] MS calcd for C 11 h 11 BrO 3 Na(M+Na) + :294.1, found: 294.1.
[0080] Synthesis of compound I2
Embodiment 3
[0082] Under nitrogen protection, 22.0g of compound 5, 29.0g of biboronic acid pinacol ester, 13.0g of potassium acetate, 3.5g of Pd(dppf)Cl were sequentially added into a 500mL reaction flask 2. CH 2 Cl 2 and 220.0 mL of 1,4-dioxane, replaced with nitrogen three times, and heated to 85°C for 6 hours.
[0083] After the central control reaction is completed, add 300.0mL water and 500.0mL methyl tert-butyl ether to the reaction bottle, stir and extract, then take the organic phase, and then extract the water phase with 200.0mL methyl tert-butyl ether once. Then, the organic phases were combined, washed once with 200.0 mL of saturated brine, and concentrated to dryness under reduced pressure at 55°C. Finally, it was purified by column chromatography (mobile phase: petroleum ether: ethyl acetate = 5:1), concentrated and dried to obtain 24.1 g of compound I2 as a yellow oil, with a purity of 94.6% and a yield of 93.0%.
[0084] 1H-NMR (400MHz, CDCl 3 )(400MHz,)δ7.96–7.91(m),7....
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More