

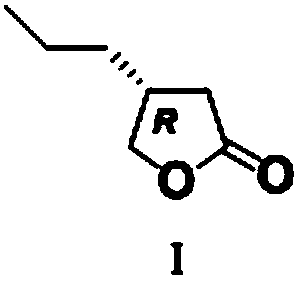
Novel butyrolactone derivative synthesizing method
A technology of butyrolactone and derivatives, which is applied in the field of new synthesis of butyrolactone derivatives, can solve problems such as difficulty in maintaining extremely high chiral purity, and achieve good stereoselectivity, reduce production costs, and low cost
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
Embodiment 1
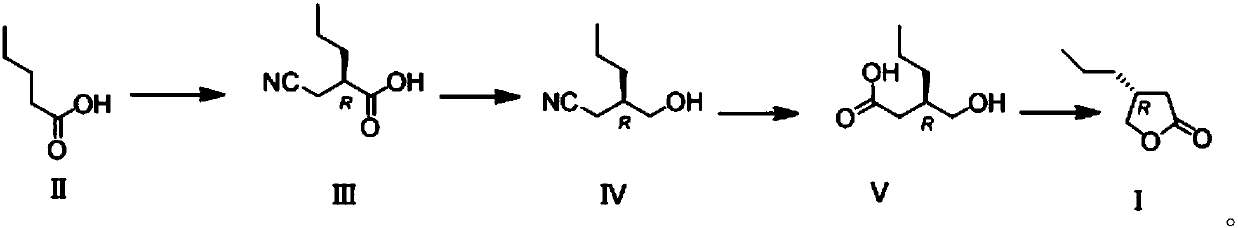
[0033] Synthesis of the compound shown in embodiment 1 formula (III)
[0034] Dissolve diisopropylamine (6.1 g, 3 equivalents) in 30 mL of ether, cool to -78°C, add 40 mL of n-butyl lithium in n-hexane (1.6M n-hexane solution, 3.2 equivalents), and at this temperature After stirring for 30 minutes, add (-)-spartenine (3.2 equivalents), then stir at this temperature for 30 minutes, add 20 mL of valeric acid ether solution (2.0 g valeric acid, 1 equivalent) represented by formula (II) , then stirred at -40°C for 1 hour, then cooled to -78°C, added 20 mL of bromoacetonitrile in ether (2.4 g bromoacetonitrile, 1 equivalent), stirred at -78°C for 2 hours, and then heated to 0 ℃, drop 50mL saturated ammonium chloride aqueous solution to terminate the reaction, wash with 75mL water, 75mL saturated brine successively, dry, spin to dry the solvent, and after column chromatography, obtain the compound shown in formula (III) (yield 56 %, e.r. value is 98:2, e.r. refers to the ratio of t...
Embodiment 2
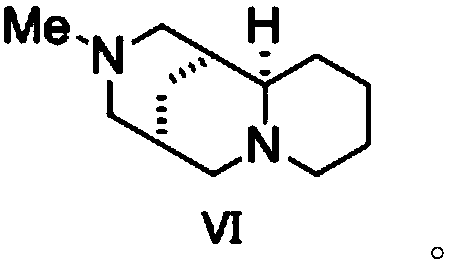
[0035] Synthesis of the compound shown in embodiment 2 formula (III)
[0036] Dissolve diisopropylamine (6.1 g, 3 equivalents) in 30 mL of ether, cool to -78°C, add 40 mL of n-butyl lithium in n-hexane (1.6M n-hexane solution, 3.2 equivalents), and at this temperature After stirring for 30 min, add the compound (3.2 equivalents) shown in formula (VI) again, then stir at this temperature for 30 min, add 20 mL of valeric acid ether solution (2.0 g valeric acid, 1 equivalent) shown in formula (II) , then stirred at -40°C for 1 hour, then cooled to -78°C, added 20 mL of bromoacetonitrile in ether (2.4 g bromoacetonitrile, 1 equivalent), stirred at -78°C for 2 hours, and then heated to 0 ℃, drop 50mL of saturated ammonium chloride aqueous solution to terminate the reaction, wash with 75mL of water, 75mL of saturated brine successively, dry, spin to dry the solvent, and after column chromatography, obtain the compound shown in formula (III) (yield 49 %, e.r. value is 96:4, e.r. ref...
Embodiment 3
[0039] The compound represented by formula (III) prepared in Example 1 (10 g, 1 equivalent) was dissolved in 200 mL of tetrahydrofuran and cooled to 0° C., and 280 mL of tetrahydrofuran borane complex (1M tetrahydrofuran, 4 equivalents) was added, 20 to 30 After stirring overnight at ℃, 100 mL of 1N (mol / L) hydrochloric acid aqueous solution was added dropwise, stirred for 1 hour, diluted with 500 mL of methyl tert-butyl ether, washed successively with 250 mL of 1N aqueous hydrochloric acid solution, 250 mL of saturated aqueous sodium bicarbonate solution, and 250 mL of saturated brine. After drying and spin-drying the solvent, the compound represented by formula (IV) was obtained (7.7 g, yield 85%). high resolution mass spectrometry (ESI + ): The theoretical value of C7H14NO+ is 128.1070, and the measured value is 128.1063.
[0040] Suspend the compound (20.3g, 1 equivalent) represented by formula (IV) in a mixture of 30mL ethanol and 10mL water, add NaOH (3g), reflux for 20...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More